Abstract
The diagnosis of seizures during the neonatal period, is difficult. Hemimegalencephaly is a rare condition with hamartomatous, characterized by unilateral enlargement of the cerebral hemisphere. The presence of resistant seizures in many patients is a poor prognostic sign. Glycine encephalopathy, also known as non-ketotic hyperglycinemia (NKH), is a rare metabolic encephalopathy, usually seen in the neonatal period. First case was a nine-day-old male patient who was hospitalized on the 6th day of follow-up because of resistant focal seizures in the right arm. He was unresponsive to midazolam infusion and phenobarbital. On magnetic resonance imaging (MRI), hemimegalencephaly was detected and the patient was referred to a epilepsy surgery center. Second case was a-34-week gestational age female newborn who was admitted to a neonatal intensive care unit. On the 21st day after hospitaiization sudden onsets of hypotonia, lethargy, apnea and hiccups were observed. Transfontaneal ultrasonography (TFUSG) showed cortical atrophy and electroencephalography (EEG) showed burst-suppression pattern. The patient was considered to have probable NKH due to the increased glycinemia concentrations in cerebrospinal fluid (CSF) and blood. The patient died in the follow-up. Neonatal seizures are rare but important as they can be accompanied by serious pathologies Rapid diagnosis and treatment can increase survival.
Keywords :
Hemimegalencephaly
, non-ketotic hyperglycinemia
, seizure
, newborn
Turkish Abstract
Yenidoğan dönemindeki nöbetlerin tanısı, çoğu zaman güçtür. Hemimegalensefali, serebral hemisferin sıklıkla bir yarısının büyümesi ile karakterize, hamartamatöz nadir bir durumdur. Dirençli nöbetlerin varlığı birçok hastada mevcuttur ve kötü prognoz belirtisidir. Non-ketotik hiperglisinemi (NKH) olarak da bilinen glisin ensefalopatisi, çoğunlukla yenidoğan döneminde görülen nadir bir metabolik ensefalopatidir. İlk vaka, dokuz günlük erkek hasta olup, takibinin 6. gününde sağ kolda başlayan fokal nöbet ve nöbetlerin midazolam infüzyonu ve fenobarbitala yanıt vermemesi nedeniyle çocuk nöroloji kliniğine konsülte edildi. Manyetik rezonans görüntülemesinde (MRG) hemimegalensefali ile uyumlu görünüm izlenen hasta epilepsi cerrahisi uygulanan bir merkeze yönlendirildi.
İkinci vaka ise 34 hafta prematürite nedeniyle yenidoğan yoğun bakım kliniğimizde takipli kız hasta olup takibinin 21. gününde aniden gelişen hipotonisite, letarji, apne ve hıçkırık nöbetleri nedeniyle konsülte edildi. Transfontanel ultrasonografisinde (TFUSG) kortikal atrofi, elektroensefalografisinde (EEG) burst-supresyon paterni izlendi. Beyin omurilik sıvısı (BOS) ve kan glisin düzeyi incelemesinde BOS glisin/kan glisin oranı artmış olarak tespit edilmesi üzerine hasta olası NKH kabul edildi. Hasta takiplerinde kaybedildi. Yenidoğan nöbetleri nadir görülmekle beraber ciddi patolojilere eşlik edebilmesi bakımından önemlidir. Hızlı tanı ve tedavi sağ kalımı arttırabilir.
Turkish Keywords :
, Hemimegalensefali
, non-ketotik hiperglisinemi
, nöbet
, yenidoğan
Introduction
Hemimegalensefali, kortikal anomalilerin eşlik ettiği, hipertrofik ve displastik serebral hemisfer ile karakterize nadir bir durumdur. Karşı serebral hemisfer genellikle basıya uğramış görünümdedir. İzole serebral bir anomali olabildiği gibi tuberoskleroz, Ito' nun unilatral hipomelanozisi ve Klippel-Trenaunay sendromu gibi durumlarla ilişkili olabilir. Tanısında magnetik rezonans görüntü (MRG) önemlidir. Klinik olarak, hayatın ilk günlerinden itibaren ortaya çıkan dirençli nöbetler ve gelişim geriliği görülür. Nöbetler birçok ilaca dirençli olup çoğunlukla epilepsi cerrahisi gerekmektedir 1, 2.
Non-ketotik hiperglisinemi (NKH), hayatın ilk günlerinden itibaren ilerleyici seyir gösteren, otozomal resesif kalıtılan, özellikle beyin omurilik sıvısı (BOS)?ta glisin artışı ile karakterize, nadir bir nörometabolik hastalıktır 3. Yenidoğan döneminde hıçkırık ve apne nöbetleri oldukça önemli olup varlığında NKH tanısını akla getirir. Altta yatan patoloji, mitokondriyal glisin parçalayıcı enzim kompleksindeki aktivitenin yetersizliğidir. Bu enzim kompleksinin tamamında, bir kısmında ya da kofaktörlerindeki yetersizlik sonucu dokularda glisin birikimi olur ve buna bağlı olarak beyin dokusu glisinin toksik etkilerine bağlı olarak etkilenir ve klinik bulgular ortaya çıkar. Çoğu hasta yaşamın ilk günlerinde kaybedilir. Elektroensefalografi (EEG)? sinde burst-supresyon paterni, hıçkırık ve apne varlığı, hipotoni ve BOS glisin/plasma glisin değerinin 0,08? in üzerinde olması genetik inceleme yapılamayan ya da mutasyon saptanmayan hastalarda tanı koymada yardımcıdır 4, 5. Biz bu çalışmada, yenidoğan nöbetlerinde nadir görülen iki farklı nöbet etyolojisini literatüre sunduk.
Case Report
Olgu-1:
Dokuz günlük erkek hasta normal vajinal yolla, gestasyonel olarak 39. haftada, 3200 gram olarak doğdu. APGAR skoru 1. ve 5. dakikada sırasıyla 7/9? du. Hastanın takiplerinde 8. saatte solunum güçlüğü gelişmesi üzerine yenidoğan yoğun bakım ünitesine yatırıldı. Fizik muayenesinde her 2 alt ve üst ekstremite simetrik hareketli, arama ve emmesi normal olarak değerlendirildi. Soy geçmişinde anne-baba 2. derece akrabaydı. Hastanın saturasyonunun oda havasında %82 olması, burun kanadı solunumu olması üzerine oksijen desteği sağlandı. Takibinin 6. gününde hastada sağ kolda ve sağ bacakta fokal nöbet varlığı izlendi (Video 1).
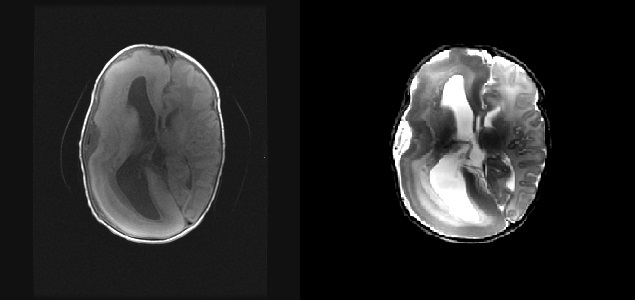
Hastaya midazolam uygulandı. Nöbetlerinin devam etmesi üzerine fenobarbital başlandı. Bakılan MRG? sinde sağ hemisfer alanında hemimegalensefali ile uyumlu görünüm izlendi (Şekil 1). Hasta epilepsi cerrahisi olan bir merkeze yönlendirildi. Hastanın ailesinden yazılı onam alındı.
Olgu-2:
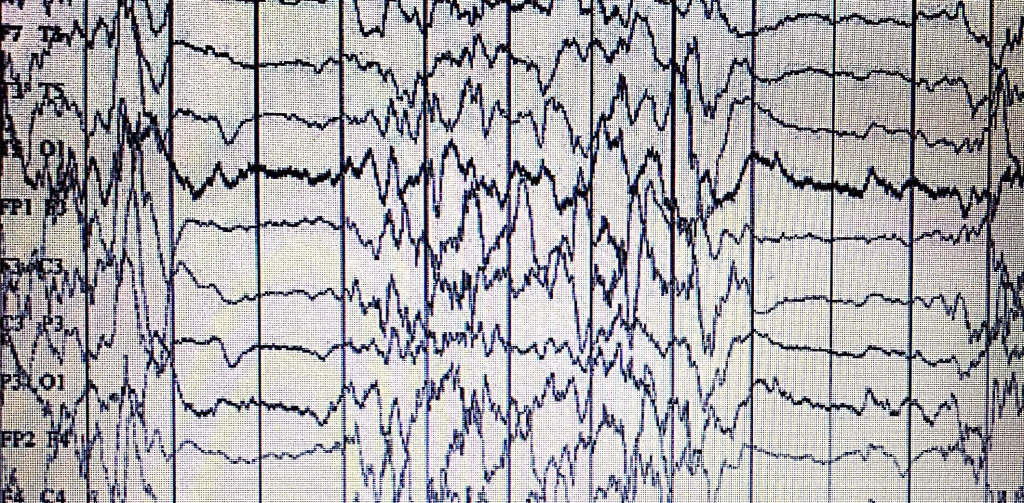
Otuz iki yaşında sağlıklı annenin 3. gebeliğinden 3. canlı doğan olarak 34 haftalık sezeryan doğum ile 2100 gram olan kız bebek doğar doğmaz ağlamadı. Burun kanadı solunumu ve her iki hemitoraksta subkostal retraksiyonları vardı. Solunum sıkıntısı nedeniyle hasta yenidoğan yoğun bakım ünitesine yatırıldı. Aile öyküsünde anne baba arasında akrabalık yoktu. Kardiyovasküler sistem muayenesinde üfürüm duyulamadı ve periferik akral siyanozu vardı. Emme, yakalama ve moro refleksleri alınabiliyordu. Tam kan ve biyokimya değerleri normal aralıktaydı. Hastanın yatışının 21. gününde solunum sıkıntısı nedeniyle nasal mekanik ventilatör desteğindeyken hipotonisite ve letarji izlendi. Hıçkırıkları ve apneleri saptandı. Kan gazında hafif solunumsal asidozu mevcuttu, amonyak değeri 365 ug/dL (0-200)? di. İdrarda keton saptanmadı. Takiplerinde miyoklonik nöbetleri gözlendi. Bakılan transfontanel ultrasonografi (TFUSG) incelemesinde kanama saptanmadı. Buna karşın yaygın serebral kortikal atrofi ile uyumlu görünüm izlendi. Bakılan EEG? sinde burst-supresyon paterni (Şekil 2) izlendi.
Bunun üzerine bakılan kan aminonasit incelemesinde serum glisin 234,50µmol/L (111-426); BOS aminoasit incelemesinde ise BOS glisin: 29,1 ?mol/L (11±4,2) olarak saptandı. Bu değerler incelendiğinde, BOS glisin/serum glisin 29,1/234,50=0,12 (Normali= <0,08) olarak saptandı. Genel durumu kötü olduğu için hastaya MR ve MR spektroskopi çekilemedi. Genetik ve enzim kompleks aktivitesinin ölçümü gibi yöntemler kliniğimizde yapılamadığından aile ile bu tetkiklerin yaptırılmasının önemi görüşüldü. Fakat aile tarafından bu tetkikler yaptırılamadı ve hasta olası NKH kabul edildi. Hastaya dekstrometorfan (5 mg/kg/gün) ve sodyum benzoat (250 mg/kg/gün) tedavisi başlandı. Buna karşın hasta yatışının 43. gününde kaybedildi. Hastanın ailesinden yazılı onam alındı.
Discussion
Hemimegalensefali, serebral hemisferin birinin anormal genişlemesi ile karakterize nadir bir beyin malformasyonudur. Klasik kliniği antiepileptik ilaçlara dirençli epilepsi, ciddi psikomotor gerilik ve hemiparezi şeklindedir. Hemimegalensefalinin etyolojisi bilinmemektedir. Genel olarak, genetik defektlerin sonucu olarak hemimegalensefalinin anormal nöronal ve glial proliferasyon veya apoptozise bağlı olduğu düşünülür 1. Hipomelanozis, İto ve tüberosklerloz gibi cilt bulgularının ön planda olduğu hastalıklara eşlik edebilir. Bazı hastalar fetal görüntüleme yöntemleriyle tanı alsa da birçok hastanın tanısında doğum sonrası başlayan nöbetler ve bu amaçla incelenen beyin MRG büyük önem taşır. Unilateral pakigri, agiri, polmikrogri, ipsilateral ventrikülomegali, ektopik gri cevher ve orta hat şiftinin varlığı önemlidir 2,6. İlk olguda yaşamının 6. gününde başlayan dirençli fokal nöbet mevcuttu. Cilt muayenesi normaldi. Hemiparezi ise yoktu. Kraniyal MRG incelendiğinde sağ hemisferde agiri, pakigri, ventrikülomegali ve orta hatta şift söz konusuydu. Antiepileptik ilaçlara dirençli nöbetleri nedeniyle hasta epilepsi cerrahisine yönlendirildi.
Non-ketotik hiperglisinemi (NKH), yenidoğan ve erken süt çocukluğu döneminde görülen, progresif, otozomal resesif geçişli, nadir bir metabolizma hastalığıdır. Sıklığı 1/250000 canlı doğumdur 3, 5. Esas metabolik kusur, piridoksal fosfat bağımlı P proteini, enzime hidrojen taşıyan H proteini, tetrahidrofolat gerektiren T proteini ve lipoamid dehidrogenaz olan L proteini olmak üzere dört özgül proteinden oluşan mitokondriyal glisin parçalayıcı enzim kompleksindeki hipoaktivitedir 4, 7. Artan glisinin beyin sapı ve spinal kord üzerindeki baskılayıcı etkisi nedeniyle beslenme güçlüğü, koma, letarji, ağır hipotoni, apne, irritabilite ve miyoklonik nöbetlerin baskın olduğu ağır ensefalopati tablosu gelişir. Beyin omurilik sıvısı glisin/ serum glisin oranı 0,08?in üzerindedir. Hıçkırık, apne ve/veya myoklonik nöbetleri olan bir yenidoğan hastada, EEG?de burst-supresyon paterni bulunması tanısaldır. Erken dönemde başlanan sodyum benzoat ve antiepileptik tedavi ise kısmen klinik fayda göstermekte olup, hızlıca glisin kısıtlı diyet başlanması gerekir 7-9. Olguların çoğu yenidoğan döneminde kaybedilir. Yenidoğan döneminden sonra yaşayan vakaların ise uzun süreli prognozu kötüdür. Hastalığa özgü bir tedavi metodu bulunmamaktadır. İkinci olguda solunum depresyonuna yol açan ağır hipotoni ve myoklonik nöbetlerin varlığı önemliydi. Keton negatifti ve EEG? de burst-supresyon paterni izlendi. Buna ek olarak BOS glisin/plasma glisin oranı artmıştı. Kesin tanıda tercih edilen lenfositlerde veya karaciğer biyopsi örneklerinde enzim kompleksininin aktivitesinin ölçümü veya moleküler düzeyde genetik mutasyonun tespiti ise mümkün olmadı.
Sonuç olarak, yenidoğan nöbetlerinin etyolojisinde birçok yapısal ve metabolik neden söz konusu olabilir. Bu makalede nöbet etyolojisinde NKH ve hemimegalensefali gibi nadir durumların olabileceği ve tanı için spesifik tetkiklerin hızlıca istenerek erken teşhisin önemine dikkat çekilmek istenmiştir. Buna ek olarak her iki durumun da prenatal tespiti mümkün olup aileler bu konuda bilgilendirilmelidir.
References
- Leventer RJ, Guerrini R, Dobyns WB. Malformations of cortical development and epilepsy. Dialogues Clin Neurosci. 2008; 10: 47?62.
- Santos AC, et al. Hemispheric dysplasia and hemimegalencephaly: imaging definitions. Childs Nerv Syst. 2014; 30: 1813?21.
- Aliefendioglu D, et al. Transient nonketotic hyperglycinemia: Two case report sandliterature review. Pediatr Neurol. 2003; 28: 151-55.
- Rossi S, et al. Early myoclonic encephalopathy and nonketotic hyperglycinemia. Pediatr Neurol. 2009; 41: 371-4.
- Madu AE, Oliver L. Non-ketotic hyperglycinaemia: case report and review of medical literature. J Matern Fetal Neonatal Med. 2013; 26: 537-9.
- Sato N, et al. Hemimegalencephaly: a study of abnormalities occurring outside the involved hemisphere. AJNR Am J Neuroradiol. 2007; 28: 678-82.
- Panayiotou E, et al. Ventilator respiratory graphic diagnosis of hiccupping in non-ketotic hyperglycinaemia. BMJ Case Rep. 2017; 9; 2017.
- Leonard JV, Morris AA. Diagnosis and early management of inborn errors of metabolism presenting around the time of birth. Acta Paediatr. 2006; 95: 6.
- Olukman Ö, et al. Nonketotic Hyperglycinemia in the Neonatal Period: Clinical Features, Diagnosis and Treatment. Tepecik Eğit Hast Derg. 2017; 27: 143-49
|