Abstract
Introduction: Glycogen storage disease type IV (GSD-IV) is a rare autosomal recessive disorder caused by deficient glycogen branching enzyme activity. This deficiency leads to the accumulation of amylopectin-like glycogen and results in variable clinical presentations, including hepatic, cardiac and neuromuscular forms.
Case presentation: We describe here a 23 years old woman with liver cirrhosis and hepatocellular carcinoma , associated with GSD-IV who was successfully treated by living donor liver transplantation in 2009. Although liver transplantation was successful , our patient died because of the cardiomyopathy and heart failure dependent to GSD-IV, approximately two years after the transplantation.
Conclusion: Although liver transplantation was successful , patients long-term follow-up is necessary to guard against neuromuscular and cardiac complications.
Keywords :
Glycogen storage disease type IV
, Liver transplantation
, Living donors
, hepatocellular carcinoma
Turkish Abstract
Giriş: Glikojen depo hastalığı tip IV (GDH-IV) nadir görülen otozomal resesif bir hastalık olup glikojen bağlayıcı enzim aktivitesinde eksiklik sonucu oluşur. Bu enzim eksikliği amilopektin benzeri glikojenin dokularda birikmesi sonucu hepatik, kardiyak ve nöromusküler formlar gibi çeşitli klinik belirtilere yol açabilmektedir.
Olgu sunumu: Biz GDH-IV ile ilişkili karaciğer sirozlu ve hepatosellüler karsinomlu (HSK) 2009 yılında canlı vericiden karaciğer transplantasyonu ile başarılı bir şekilde tedavi edilen 23 yaşında kadın hastayı sunduk. Karaciğer transplantasyonu başarılı olmasına rağmen hastamız transplantasyondan yaklaşık 2 yıl sonra GDH-IV e bağlı kardiyomyopati ve kalp yetmezliği nedeni ile öldü.
Sonuç: Her ne kadar karaciğer transplantasyonu başarılı olsada hastaların kardiyak ve nöromüsküler komplikasyonların takibi ve önlenebilmesi için uzun dönem takibi gerekmektedir.
Turkish Keywords :
, Glikojen depo hastalığı tip IV
, karaciğer transplantasyonu
, Canlı donör
, hepatosellüler karsinom
Introduction
Glycogen storage disease type IV (GSD-IV) or ( Andersen Disease, amylopectinosis) is a rare autosomal recessive disorder caused by deficient glycogen branching enzyme (GBE) activity 1. It has been diagnosed by measuring glycogen branching enzymes in individual tissues 2. This deficiency leads to the accumulation of amylopectin-like glycogen (polyglucosan) and results in variable clinical presentations, including hepatic, cardiac and neuromuscular forms 3. Fatal hepatic complications usually occur by the age of 2?4 years, but in exceptional cases involvement in other organ systems may be prominent mortality factors 4,5. Liver transplantation for this disease was first attempted in 1972, but the recipient died approximately three months later after uncontrolled rejection of his first liver and attempted retransplantation 6. We describe here a 23 years old woman with liver cirrhosis and hepatocellular carcinoma, associated with GSD-IV who was successfully treated by living donor liver transplantation in 2009.
Case Report
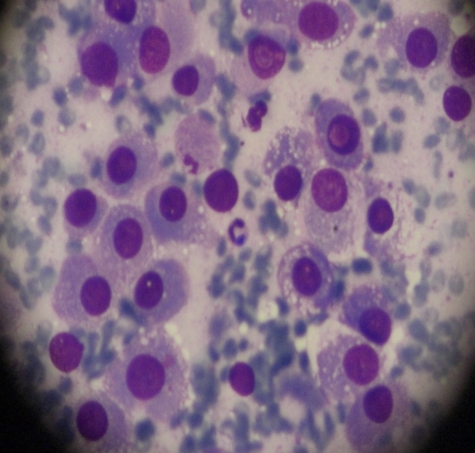
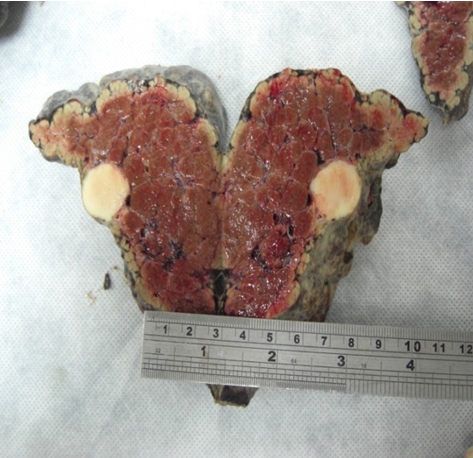
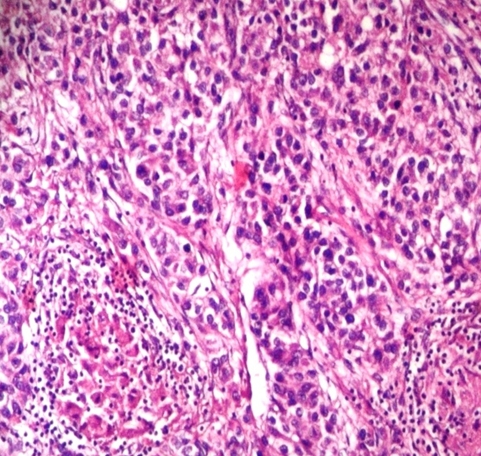
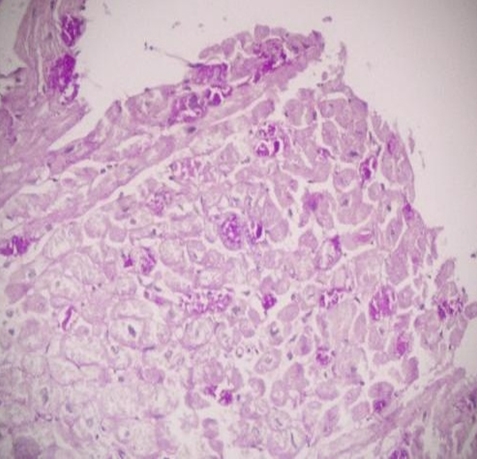
A 23 years old woman who have a diagnosis of GSD-IV for past five years, initially visited our hospital with hepatomegaly. Her blood pressure was 100/60 mmHg, heart rate 75 beats per min, respiratory rate 14 breaths per min, and body temperature was 36.5oC. On physical examination, liver was firm and palpable by two fingerbreadth below the rib and abdomen was distended without ascites. Neurologic examination showed no definite abnormality and muscle tone was normal. Laboratory tests showed hemoglobin 11.8 g/dL, platelet count 63,000/mm3, white blood cell count 3,500/mm3 ,total protein 7.0 g/dL,albumin 4.2 g/dL, alanine aminotransferase 22 IU/L, aspartate aminotransferase 27 IU/L, total bilirubin 0.8 mg/dL, direct bilirubin 0.1 mg/dL, gamma-glutamyltranspeptidase 44 IU/L, alkaline phosphatase 45 IU/L, prothrombin time 35.3% (1.22 INR), and activated partial thrombin time 31 sec. Serologic markers for hepatitis A virus, hepatitis B virus and hepatitis C virus were negative. A chest X-ray showed mild cardiomegaly but had no clinical evidence of cardiac dysfunction who were tested with echocardiogram or cardiac catheterization had a normal cardiac function before the liver transplantation. Computerized tomography ( CT) described a hypervascular mass in the right hepatic lobe (2.5 × 2.3 ×1.9 cm) which seems like hepatocellular carcinoma(HCC) and described portovenous shunts. An ultrasound guided fine needle aspiration of mass showed atypical hepatocytes and HCC was diagnosed by FNA (Figure 1a). She underwent living donor liver transplantation from her sister, a heterozygous donor. The transplant was uneventful. The explanted liver, which measured 17×13×8 cm and weighed 491 gm was diffusely nodular with a brown color. Cut surface revealed round, well-demarcated, smooth, solid nodule, 23 mm in diameter (Figure 1b). Microscopic examination revealed variable-sized regenerating nodules, supporting the diagnosis of cirrhosis. The intracytoplasmic globuls were strongly positive on periodic acid-Schiff (PAS) staining (Figure 1c). The histopathological examination for the resected specimen yielded the final diagnosis of the mass which was 2.5x2.2x2 cm in the right hepatic lobe was hepatocellular carcinoma (Figure 1d). Following liver transplantation, the patient was treated with standard immunosuppressive therapy. Six months after transplantation, she is well, with normal liver function.
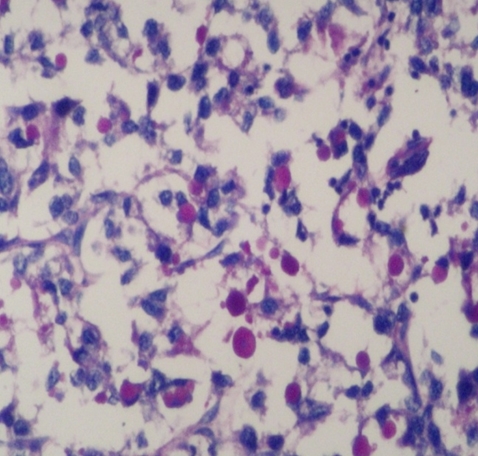
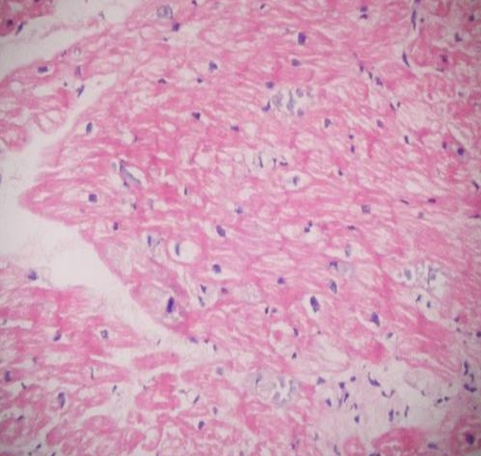
Due to cardiac symptoms, myocardial biopsy obtained 18 months after liver transplantation. On histopathological examination of heart, there was marked vacuolisation of myocytes with few bizzare nuclei seen (Figure 2a). A heart biopsy demonstrated Periodic Acid-Schiff (PAS) positive, diastase resistant, coarsely clumped, granular and fibrillary material typicall of amylopectin (Fig.ure 2b). She died because of the cardiomyopathy and heart failure dependent to GSD-IV, two years after transplantation.
Discussion
GSD IV is characterized by a remarkable heterogeneity in the clinical and biochemical expression of the disease. Indeed, mutations in the same GBE gene are responsible for presentations as different as the classical hepatic form, hydrops fetalis with arthrogryposis, severe neuromuscular congenital form, and an adult neuromyopathy named polyglucosan body disease 7,8. Glycogen storage diseases are more than 12 types and they are classified based on the enzyme deficiency and affected tissue. Our patient presented with features of milder nonprogressive hepatic forms of GSD-IV which was less frequently seen 7. GSD-IV, various neuromuscular forms are known, which differ according to age at onset and severity and which present with symptoms of myopathy, cardiopathy, and central and peripheral nervous system dysfunction (8). There have been no neuromuscular complications in our patients. Our patient showed mild cardiomegaly on chest X-ray but cardiac function tests and echocardiography were within normal limits. Diagnosis of GSD-IV includes the histologic detection of abnormal glycogen storage and the biochemical measurement of deficient GBE activity, postnatally in red blood cells, leukocytes, hepatocytes, and cultured fibroblasts, and/or prenatally in amniotic fluid and chorionic villus cells 9,10. Liver transplant is the only therapeutic option for the classical form of GSD IV characterized by early liver failure. Because amylopectin-like material is not soluble and GBE is present in other tissues, including the heart or the muscles, extrahepatic progression of the disease is a potential risk for these patients. The first successful liver transplantation for GSD-IV was performed in 1984 11. The absence of amylopectin deposition in the liver grafts was expected, but the independed from neuromuscular or cardiac morbidity associated with extrahepatic amylopectin deposits was more note worthy. Our patient had no clinical evidence of pretransplant cardiac dysfunction who were tested with echocardiogram or cardiac catheterization had a normal cardiac function before the liver transplantation. Extrahepatic deposits of polyglucosan have been reported to be resorbed through the migration of donor cells from the liver allograft 12, although our patient died from cardiomyopathy, two years after liver transplantation. In a study the patients received transplants from living, related, heterozygous donors, as did our patient, and no mortality or morbidity related to heterozygosity has been observed 13. However, of the 7 patients described by Selby et al 14, 2 had cardiac amylopectin-like deposits only a few weeks after the LT, which was probably already present before the procedure, and none had a progressive cardiomyopathy.
We report a patient diagnosed with GSD-IV and hepatocellular carcinoma who underwent successful living donor liver transplantation from heterozygous donor. Our experience in LT for GSD IV ,patient who had a fatal course because of extrahepatic posttransplant progression of the disease. Two patients with GSD IV have been previously reported to have died of cardiomyopathy with amylopectin- like material deposits in myocytes two years after LT 15. Although the cause of death for our patients was heart failure with a massive accumulation of amylopectin-like material and reduced myofibrils in cardiomyocytes, other organs might also be infiltrated.
Conclusion:
Although liver transplantation was successful , patient died because of the cardiomyopathy and heart failure dependent to GSD-IV, approximately two years after the transplantation. Our observations show that LT may not alter the extrahepatic progression of GSD IV. Functional evaluation of the heart does not provide any predicting factor in this situation. Therefore, although liver transplantation was successful in the patient, long-term follow-up is necessary to guard against neuromuscular and cardiac complications.
References
- Andersen DH. Familial cirrhosis of the liver with storage of abnormal glycogen. Lab Invest. 1956;5:11-20.
- Lee KY, Seo KH, Lee HK, Kim JW. Glycogen storage disease type IV: a case report. J Korean Med Sci. 1998;13: 211-4.
- Moses SW, Parvari R. The variable presentations of glycogen storage disease type IV: a review of clinical, enzymatic and molecular studies. Curr Mol Med. 2002;2:177-88.
- Greene GM, et al. Juvenile polysaccharidosis with cardioskeletal myopathy. Arch Pathol Lab Med. 1987;111:977?82. [PubMed: 2957974]
- Hers HG, Van Hoof F, De Barsy T. Glycogen storage diseases. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic Basis of İnherited Disease. 6th edn. New York: McGraw Hill; 1989. p. 425-53.
- Starzl TE, et al. Fifteen years of clinical liver transplantation. Gastroenterology. 1979;77:375?88. [PubMed: 376395]
- Cori GT, Cori CF. Glucose-6-phosphatase in the liver in glycogen storage disease. J Biol Chem. 1952;199:661?7. [PubMed: 13022673]
- Bruno C, et al. Clinical and genetic heterogeneity of branching enzyme deficiency (glycogenosis type IV). Neurology. 2004;63:1053-8.
- Brown BI, Brown DH. Lack of an alpha-1,4-glucan: alpha- 1,4-glucan 6-glycosyl transferase in a case of type IV glycogenosis. Proc Natl Acad Sci (U S A ). 1966;56:725-9.
- Thon VJ, Khalil M, Cannon JF. Isolation of human glycogen branching enzyme cDNAs by screening complementation in yeast. J Biol Chem. 1993;268:7509-13.
- Selby R, et al. Liver transplantation for type I and type IV glycogen storage disease. Eur J Pediatr. 1993;152:S71-S76.
- Starzl TE, et al. Chimerism after liver transplantation for type IV glycogen storage disease and type 1 Gaucher's disease. N Engl J Med. 1993;328: 745-9.
- Morioka D, et al. Living donor liver transplantation for pediatric patients with inheritable metabolic disorders. Am J Transplant. 2005;5:2754-63.
- Selby R, et al. Liver transplantation for type IV glycogen storage disease. N Engl J Med. 1991;324:39?42.
- Rosenthal P, et al. Failure of liver transplantation to diminish cardiac deposits of amylopectin and leukocyte inclusions in type IV glycogen storage disease. Liver Transpl Surg. 1995;1:373?6.
Information Presentation
This paper was presented as a poster at the 24th Conference of APASL, 2015, 12-15 March, 2015, Istanbul.
|