Abstract
A 34-year-old woman during her second pregnancy, applied to our perinatology clinic for triple test. Posterior encephalocele, oligohydramnios, polycystic kidneys and polydactyly were identified in the ultrasound examination. Meckel-Gruber syndrome (MGS) was considered. A detailed briefing was given to the pregnant woman and termination option was introduced. The pregnancy was terminated at 18 weeks of gestation with the choice of the family. Autopsy results also confirmed the diagnosis.
MGS is a syndrome that should be differentiated from trisomy 13 in the perinatal period and the final diagnosis is confirmed by autopsy after termination. The hereditary process should be explained to the family. We think that early diagnosis and termination of this fatal syndrome would be very helpful for reducing maternal physical and psychological trauma.
Here, we discussed this rare and fatal syndrome in the light of literature.
Keywords :
Meckel-Gruber syndrome
, Prenatal diagnosis
Turkish Abstract
Kliniğimizin Riskli Gebelik Polikliniğine üçlü test yaptırmak için ilk kez başvuran ikinci çocuğuna gebe 34 yaşındaki hastamıza yapılan ultrasonografik muayenede; fetusta, posterior ensefalosel, oligohidramnios, polikistik böbrekler ve polidaktili tespit edildi. Meckel Gruber Sendromu (MGS) düşünülen gebeye detaylı bilgilendirme yapıldı ve terminasyon seçeneği sunuldu. Terminasyon isteyen ailenin gebeliği 18. haftada sonlandırıldı. Otopsi sonuçları da tanıyı doğruladı.
Biz, bu yazımızda, nadir görülen ve ölümcül olan MGS?nu literatür eşliğinde tartıştık.
Turkish Keywords :
, Meckel-Gruber sendromu
, Prenatal tanı
Introduction
Meckel Gruber sendromu (MGS), perinatal dönemde trizomi 13?den ayrımı gereken ve kesin tanısı terminasyon sonrası otopsi ile konulan bir sendromdur. İlk olarak 1822 yılında Johann Freidrich Meckel tarafından tanımlanmıştır. Daha sonra 1934 yılında Gruber tarafından yeniden değerlendirilip düzenlenmiştir. Literatürde Dysencephalia Splanchnocystica olarak da tanımlanmaktadır 1.
MGS, otosomal resessif (OR) geçişlidir ve sorumlu genin 17q 21-24?de lokalize olduğu belirlenmiştir. Standart karyotip incelemeler normal olup DNA düzeyinde incelemeler yapılması gerekmektedir 2.
Aileye genetik geçiş ile ilgili bilgi verilmesi gerekir. Ölümcül olan sendromda erken tanı ve terminasyonun, annenin fiziksel ve psikolojik travmasının azaltılmasında faydalı olacağı düşüncesindeyiz.
Amacımız, nadir görülen ve ölümcül bir durum olan MGS?unu literatür eşliğinde tartışmaktır.
Case Report
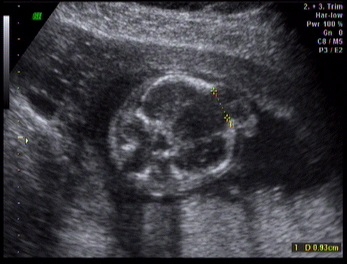
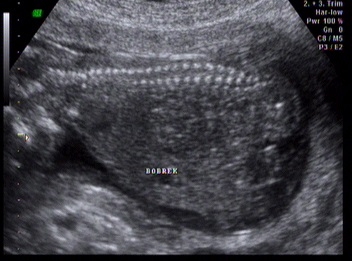
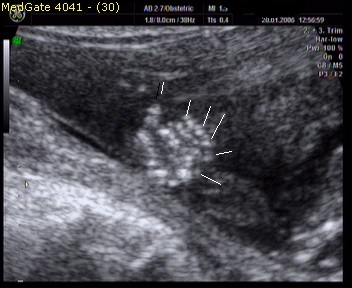
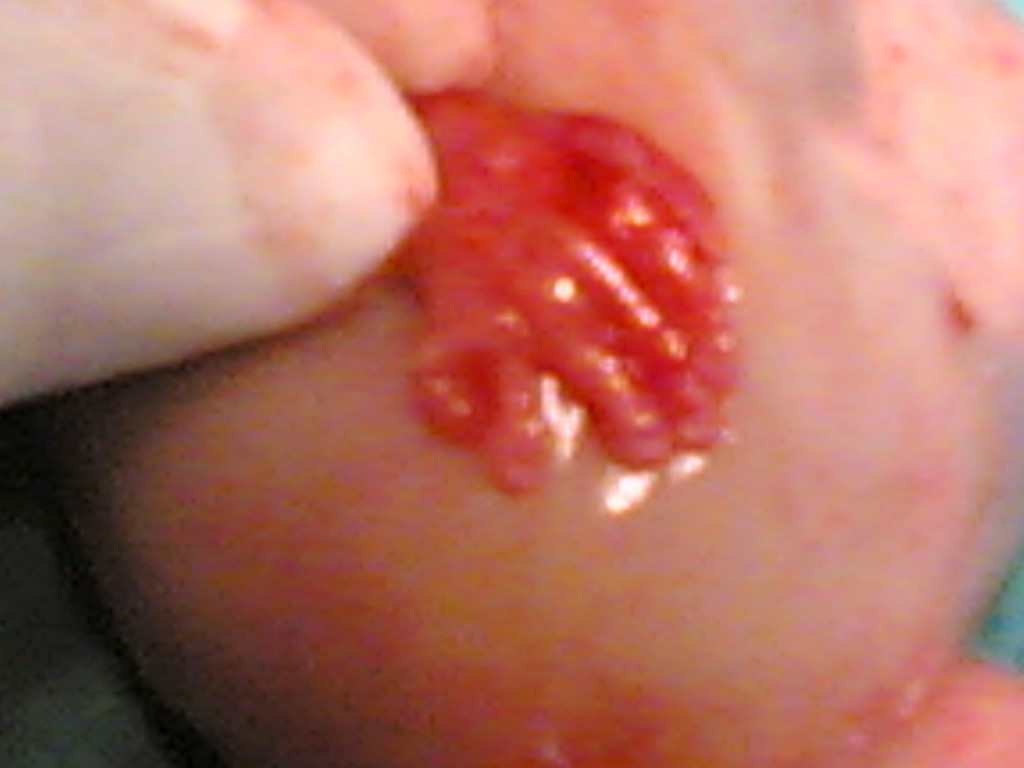
Kliniğimiz Riskli Gebelik polikliniğine üçlü test yaptırmak için ilk kez başvuran 34 yaşındaki 2. çocuğuna gebe hastamıza yapılan ultrasonografik muayenede; fetusta, posterior ensefalosel (Şekil 1), oligohidramnios, polikistik böbrekler (Şekil 2) ve polidaktili (Şekil 3) tespit edildi.
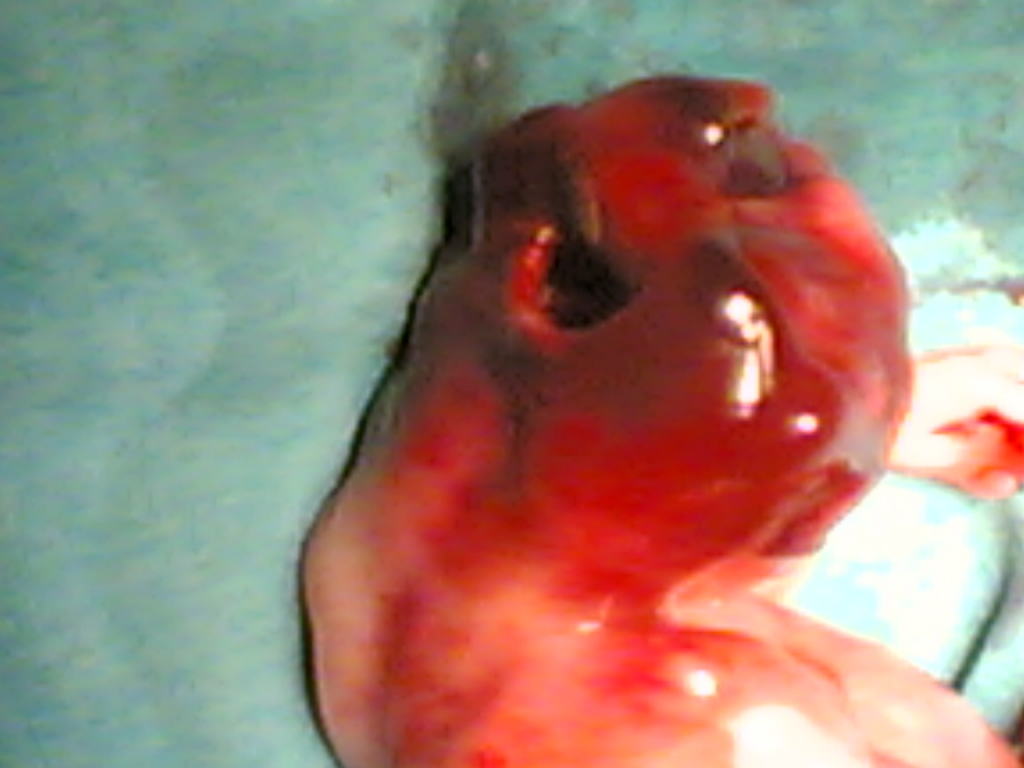
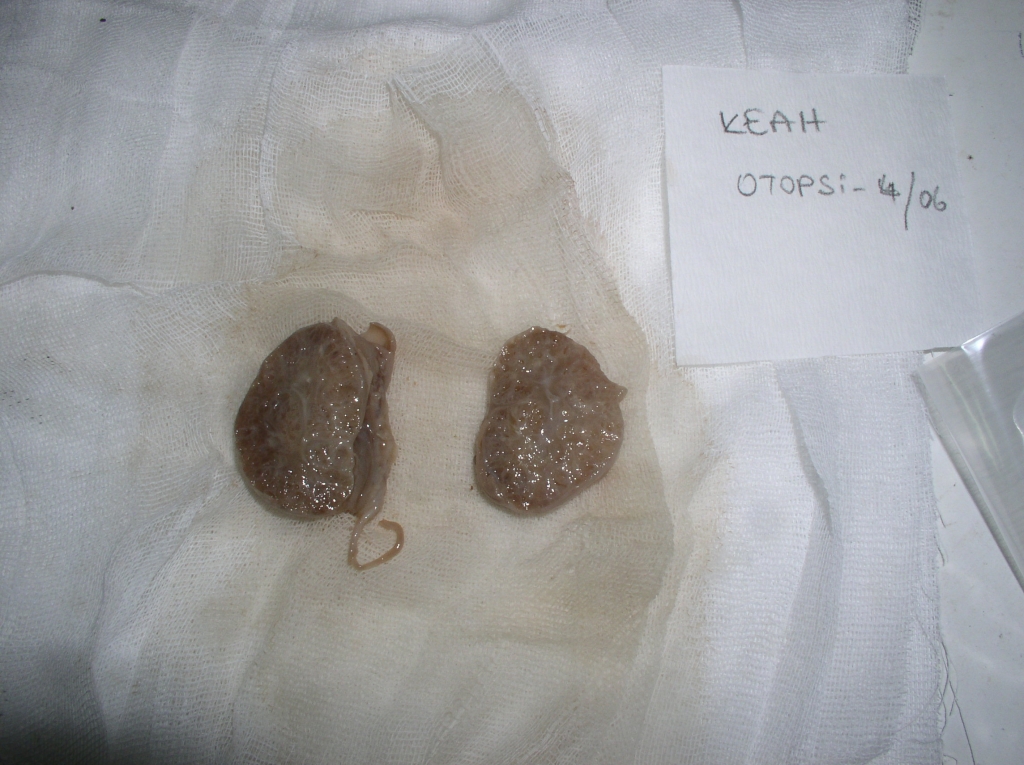
Gebenin öz ve soygeçmişinde bir özellik yoktu ve ilk çocuğu sağlıklı idi. MGS düşünülen gebeye sendrom hakkında detaylı bilgilendirme yapıldı ve aileye terminasyon seçeneği sunuldu. Terminasyon isteyen ailenin gebeliği 18. haftada sonlandırıldı. Otopsi sonuçları da tanıyı destekledi (Şekil 4?7).
Discussion
Ölümcül bir hastalık olan MGS?nin , moleküler genetik incelemeler ile otozigosite haritalaması kullanılarak 3 ayrı lokusu tanımlanmıştır. MGS1 lokusu 17q21-24, MGS2 lokusu 11q13, MGS3 lokusu 8q24 üzerindedir. MGS3 bağlantılı ailelerde polidaktili ve ensefalosel MGS1 ve MGS2 bağlantılı benzerlerinden farklı olarak daha nadir görülür 3. Bu farklı kromozomlar fenotipik değişkenliği açıklamaktadır. Aynı soydan gelen ailelerde insidansında artış olduğu da belirtilmiştir 4. Bizim vakamızda ailesel öyküde hiçbir özellik bulunamamıştır.
MGS?da ölüm inutero veya erken postnatal dönemde gerçekleşebilir. Oligohidramnios pulmoner hipoplaziye neden olarak ölümün en önemli sebebidir. Doğumdan sonra ilk birkaç saat içinde ölüm görülebilir 5. Nadir vakalarda düşük yaşam kalitesi ile birkaç aylık yaşam görülebilir 6. Bu nedenle gebelikte belirlenmesi durumunda gebeliğin sonlandırılması önerilmektedir ve biz de aileye bu doğrultuda bilgilendirme yaptık.
Görülme sıklığı 12,000?140,000 ?de birdir, her iki cinste görülebilir 7. Ancak farklı ırklarda bu oranlar değişiklik gösterebilmektedir.
Polikistik böbrek %100, oksipital ensefalosel %90 ve postaksiyal polidaktili %83 oranında görülmektedir ve bu üç anomali MGS?nin klasik triadını oluşturmaktadır. Klasik triadın %57 hastada görüldüğü belirtilmiştir. Bizim hastamızda da klasik triad mevcut idi. Ancak tanı için bu 3 bulgudan 2?sinin bulunması yeterli kabul edilmektedir 8. Bizim vakamızda her üç bulgu da izlenmiştir.
Böbrekler 10?20 kata kadar büyümüş olabilir. Genellikle ekojeniktirler ve geniş makroskobik kistler de içerebilirler. Genişlemiş böbrekler karın çevresinde belirgin genişlemeye ve abdomen çevresi ölçümünde belirgin fazlalaşmaya neden olabilirler 4.
Ensefaloselin boyutu değişkendir ve yerleşim yeri oksipital alandadır. Mikrosefali eşlik edebilmektedir 4.
Postaksiyal polidaktilide fazladan görülen parmak küçük veya açılanmış olabilir. Genellikle her 4 ekstremiteyi de benzer olarak etkiler ancak bu belirtisi triadın en çok değişkenlik gösteren belirtisidir 4.
Bu anomaliler dışında sendroma eşlik eden daha nadir görülen birçok yapısal anomali olduğu da belirlenmiştir. Santral sinir sistemi (SSS) anomalileri içinde; mikrosefali, hidrosefali, Dandy Walker malformasyonu, Arnold Chiari malformasyonu, korpus kallosum agenezisi, serebral hipoplazi, genitoüriner anomaliler içinde; ambiguous genitalia, renal agenezi, renal hipoplazi, displastik böbrekler, üreteral duplikasyon, kriptorşidizm, mesane hipoplazisi, karaciğerde; duktal plate malformasyonu, fibrotik değişiklikler ve safra duktus proliferasyonu, pankreasda; kistler ve fibrotik değişiklikler, kalp defektleri olarak; ASD, VSD, PDA, aort koarktasyonu, pulmoner stenoz anomalileri, iskelet sisteminde; polidaktili, syndaktili, klinodaktili, kısa ekstremite, uzun kemiklerde eğilme, club foot, simian çizgisi, yarık damak dudak, mikrognati, göz anomalileri; mikroftalmi, surrenal hipoplazi ve oligohidramnios, anhidramnios gibi birçok farklı patolojinin eşlik edebildiği belirtilmiştir 1-8. Bizim vakamızda oligohidramnios dışında ilave patoloji izlenmemiştir.
Kesin tanı otopsi incelemesi ile konulur. Karyotip tayininin normal olması tanıyı destekleyen bulgudur. Özel bir biyokimyasal belirteç veya kromozomal belirteç olmadığı belirlenmiştir. Prenatal dönemde tanı, ultrasonografi belirteçlerinin saptanması ve kromozom incelemesinin normal olması ile konulur. Kesin tanı için kromozom incelemesi ve postabortal veya postnatal otopsi gerekmektedir. Vakamızda bu tanısal uygulamaya göre hareket edilmiştir.
Prenatal dönemde tanı en erken 10.haftada fetoskopi ile konulabilir. Ancak rutin bir işlem olmadığı için sadece özgeçmişinde MGS öyküsü olanlarda kullanılması düşünülebilir. Quintero ve ark. 11. haftada MGS?li fetüsü transabdominal embryofetoskopi ile tanımışlardır 9. Sonraki gebeliklerinde bu tanı yöntemlerinin de kullanılabileceği bilgisi aile ile paylaşılmıştır.
Ultrasonografi incelemeleri ile 11?14. haftalar arasında ense kalınlığı (nuchal translucency (NT) ve baş-popo uzunluğu (crown-lump lenght (CRL)) ölçümleri yapılırken fetal değerlendirme sırasında saptanabilirler. Blaas ve ark. 3 boyutlu ultrasonografinin bu tür anomalileri saptamada etkili olduğunu düşünmüşler ve çeşitli SSS anomalilerini saptamışlardır 10.
Barisic ve ark. 1990?2011 yılları arasındaki 34 Avrupa ülkesinden 191 MGS vakasını incelemişlerdir 11. 145 vaka (%75,9) termine edilmiş, 13 vaka (%6,8) ölmüş, 33 vaka (%17.3) canlı doğmuştur. Vakalarının %90?ının 14,3+/-2,6 haftalarda saptandığını belirlemişlerdir 11.
Paelkar ve ark. araştırmasında canlı doğan iki vakadan birinin 18 ay diğerinin ise 28 ay yaşadıklarını bildirmiştir 12.
Alfa feto protein değerlendirmesi spesifik değildir ancak tanıya yardımcı olabilmektedir ve bu hormonun düzeyi MGS hastalarındaki ensefalosel nedeniyle yükselmiş olarak saptanabilir. Trizomi 13?den ayırıcı tanıda anlamlı olabilir. Ancak membranla kaplı olanlarda AFP seviyesi normal olarak da saptanabilir 13.
Ayırıcı tanıda, trizomi 13?de ayrıca %5-30 kadarında kistik böbrekler olabilmektedir. Bunun yanında orta hat SSS anomalileri veya holoprosensefalinin de görülebildiği belirtilmiştir. Trizomi 13?de de ensefalosel bildirilmiştir ancak nadirdir ve farklı olarak rahim içi gelişme geriliği (IUGG), omfalosel, rocker bottom foot gibi anomaliler de eşlik edebilir 4. Bizim vakamızda da ayırıcı tanıda diğer tüm belirteçlere bakılmıştır. Oligohidramnios dışında ilave anomali izlenmemiştir.
Prenatal dönemde ultrasonda trizomi 13 dışında Smith Lemni Opitz sendromundan da ayrılması gerekebilir. Smith Lemni Opitz sendromunda ise SSS malformasyonu, genitoüriner sistem malformasyonları, polidaktili ve karaciğer değişiklikleri görülebilmektedir, bu sendrom da OR geçişlidir 14.
Prenatal dönemde Meckel Gruber sendromu tanısı konanlarda terminasyon seçeneği sunulur. Ancak gebeliğin devamını isteyenlerde fetal monitorizasyon ve sezaryen önerilmemektedir 4. Miad doğumlarda genişlemiş karın çevresi nedeniyle abdominal distosi gelişebilir 4.
Oligohidramnios nedeniyle ideal bir değerlendirme yapılamayan gebeliklerde manyetik rezonans inceleme yapılabileceği de belirtilmiştir 4. Bizim vakamızda ultrasonografi ile yeterli değerlendirme yapılabilmiştir, ilave bir tanı metoduna ihtiyaç duyulmamıştır.
Sonuç
MGS?nin perinatal dönemde trizomi 13?den ayırıcı tanısının yapılması faydalı olacaktır. Çünkü trizomi 13?ün tekrarlama riski %1 iken MGS?nin tekrar görülme riski %25?tir. MGS tanısı alan ailelerle sonraki gebeliklerinde embryoskopi yöntemiyle erken tanı sağlanabileceği paylaşılmalı ve isteyen çiftler bu teknik konusunda deneyimli merkezlere yönlendirilmelidirler. Ölümcül olan bu sendromun erken tanı ve erken müdahalesi sayesinde maternal fiziksel ve psikolojik travmanın azaltılmasında faydalı olunacağı düşüncesindeyiz.
References
- Kılınç N. ve ark., Meckel-Gruber Sendromu (Bir Otopsi Olgusu). Perinatoloji Dergisi. 2002;10(4):4.
- Salonen R, Paavola P. Meckel syndrome. J Med Genet. 1998;35:497?501.
- Sepulveda W, et al. Diagnosis of the Meckel Gruber Syndrome at eleven to fourteen weeks?gestation. Am J Obstet Gynecol.1997;176:316-9.
- Woodward PJ, et al. Diagnostic imaging obstetrics. Meckel Gruber Syndrome. Utah:Amirsys, 2005. p.15/22-25.
- Alam A. Meckel Gruber Syndrome: Second trimester diagnosis of a case in a non-consanguineous marriage. Pak J Med Sci. 2013;29(1):234-6.
- Arthur CF, Eugene CT, Wesley L, Frank AM, Roberto R. Atrioventricular septal defects. In: Sonography in Obstetrics and Gynecology: Principles and Practice. 7th edition. Atrioventriculer septal defects. New York: McGraw-Hill, 2011. p.629-630.
- Myageri A.et al. Meckel Gruber syndrome: report of two cases with review of literature. J Family Med Prim Care. 2013;2(1):106-8.
- Sayhan S ve ark. Meckel-Gruber Sendromu. Perinatoloji Dergisi. 2004;12(2):99-101.
- Quintero RA, et al. Transabdominal thin-gauge embryofetoscopy: a technique for early prenatal diagnosis and its use in the diagnosis of a case of Meckel-Gruber syndrome. Am J Obstet Gynecol. 1993;168(5):1552-7.
- Blaas HG Sonoembryology and early prenatal diagnosis of neural anomalies. Prenat Diagn. 2009;29(4):312-25.
- Barisic I. Meckel-Gruber Syndrome: a population-based study on prevalence, prenatal diagnosis, clinical features, and survival in Europe. Eur J Hum Genet. 2014 Sep 3. doi: 10.1038/ejhg.2014.174. [Epub ahead of print]
- Parelkar SV Meckel-Gruber syndrome: A rare and lethal anomaly with review of literature. J Pediatr Neurosci. 2013;8(2):154-7.
- Gazioğlu N, et al. Meckel-Gruber syndrome. Childs Nerv Syst. 1998;143:142-5.
- Moerman P, et al. The pathology of trisomy 13: a study of 12 cases. Hum Genet. 1988;80:349-56.
Information Presentation
2006 yılında Ulusal Perinatoloji Kongresi?nde İstanbul Harbiye Askeri Müzesi?nde poster sunumu olarak sunulmuştur.
|