Abstract
The Mayer-Rokitansky-Kuster-Hauser syndrome (MRKHS) is characterized by congenital aplasia of the uterus and upper part (2/3) of the vagina in women showing normal development of secondary sexual characteristics with a normal 46, XX karyotype. The combination of MRKHS with renal anomalies and cervicothoracic dyspla¬sia is known as MURCS association (Mullerian aplasia, Renal dyspla¬sia and Cervical Somite Anomalies). A 5 year old girl, presented with complaints of prematüre pubarche, identified because of the shortness of the thumb in her physical examination, the further investigation it turned out to be MRKHS.
Keywords :
Mayer-Rokitansky-Kuster-Hauser syndrome
, Premetüre pubarche
, Anomaly of finger
, Vaginal atresia
Turkish Abstract
Mayer-Rokitansky-Kuster-Hauser sendromu (MRKHS), normal sekonder cinsiyet karakteri mevcut olan ve 46,XX karyotipe sahip kadınlarda, uterus ve üst 2/3 vajinanın yokluğuna denilmektedir. MRKHS?nin renal anomaliler ve servikotorasik displazi ile birlikteliği Müllerian kanal aplazisi, Renal displazi ve Servikal Somit Anomalisi (MURCS) olarak bilinmektedir. Pubik kıllanma şikayeti ile başvuran 5 yaşında kız olgunun fizik muayenesinde tespit edilen baş parmakta kısalık nedeniyle yapılan ileri tetkiklerde MRKHS olduğu tespit edilmiştir.
Turkish Keywords :
, Mayer-Rokitansky-Küster-Hauser sendromu
, Pubik kıllanma
, Parmak anomalisi
, Vajinal atrezi
Introduction
Müllerian agenezi ve Müllerian kanaldan orijin alan organların yokluğu Mayer-Rokitansky-Küster-Hauser sendromu (MRKHS) olarak tanımlanır. Bu sendrom 2 tipdir: tip I (izole) veya Rokitansky (OMIM 277000) ve tip II veya Müllerian kanal aplazisi, Renal displazi ve Servikal somite anomalisi (MURCS) (OMIM 601076) 2?dir. Genellikle sporadik ortaya çıkar ve eşlik eden anomaliler; servikotorasik vertebral defektler, vajina yokluğu, uterus hipoplazisi veya yokluğu, renal agenezi veya ektopidir. Bu hastalar sıklıkla normal ikincil cinsiyet bulgularına sahip olup, primer amenore veya infertilite nedeniyle başvurmaktadır. Tek taraflı üst ekstremite malformasyonu %26 vakada tanımlanmıştır. Erkek hastalarda da tanımlanmış olup azospermi, renalanomali, servikotorasik omurga defekti (ARCS) olarak kısaltılmıştır 1. Bu yazıda, 5 yaş dokuz aylık, genital tüylenme yakınması ile getirilen, yapılan hormonal incelemeleri sonucunda "prematür adrenarş" tanısı alan kız hasta sunulmaktadır. Prematür adrenarş tanısından bağımsız olarak fizik muayenede ve el grafisinde baş parmak hipoplazisi saptanması üzerine eşlik edebilecek renal anomaliler için incelenmiş ve renal agenezi saptanmıştır. Sonrasında, üriner sistem anomalisine eşlik edebilecek genital sistem anomalisi açısından incelenerek Müllerian agenezi saptanmıştır. Bu yazımızda, prematur genital kıllanma ile başvuran ve yukarıda belirtilen bulgularla MRKHS tanısı alan 5 yaşındaki hasta sunulmuştur.
Case Report
Beş yaş dokuz aylık kız hasta, bir yıl önce başlayan pubik kıllanma şikayeti ile getirildi. Fizik muayenesinde ağırlığı 20 kg (50p), boyu110 cm (25-50p), nabız:100/dk, solunum:16/dk, kan basıncı:110/70 mmHg idi. Baş ve boyun muayenesinde saç ve saçlı deri doğaldı, boyun hareketlerinde kısıtlılık yoktu, kısa boyun ve yelelenme yoktu (Şekil 1a).
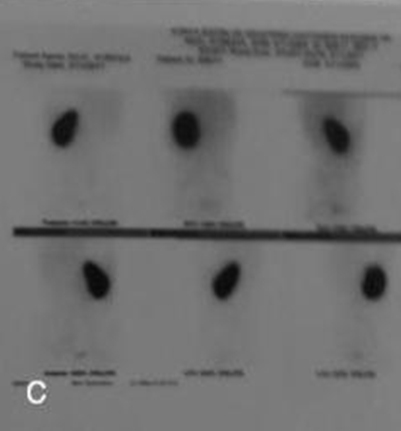
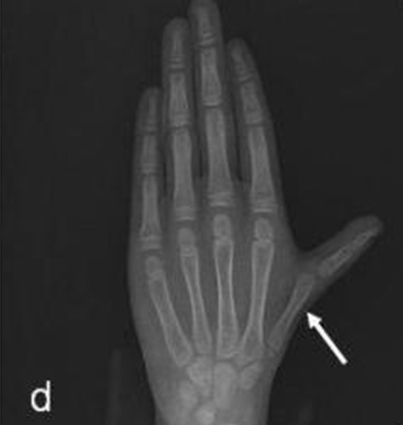
Ekstremite muayenesinde sol el birinci metakarpal kemiğin kısa olduğu tespit edildi. Hastanın sol el bilek grafisinde de birinci falanksta kemik deformitesi mevcut olduğu görüldü (Şekil 1b,, 1c).
Pubertal değerlendirmede, meme gelişimi Tanner evre 1, pubik tüylenme evre 2 idi. Sistemik muayene bulguları normaldi.Hemogram, biyokimya ve hormonal değerler ( FSH: 0,88 mIU/ml, LH: 0,01 mIU/ml, östradiol: 0,01 pg/ml, sT4: 1,5 ng/dl, TSH: 1,83 µIU/ml, testosteron: 33,82 ng/dl, DHEAS: 140 µg/dl, kortizol: 15,48 µg/dl, ACTH, Kortizol, 17OH Progesteron) normaldi. ACTH testinde uyarılmış kortizol, 17 OH progesteron ve DHEAS düzeyleri normaldi. Kemik yaşı 4 yaş ile uyumlu idi.
Parmak anomalisi olan hastanın eşlik eden renal anomali olup olmadığını tespit etmek için yapılan renal ultrasonografisinde tek taraflı renal agenezi tespit edildi. Sol böbrek ise normal olarak değerlendirildi. Renal sintigrafide tek, normal fonksiyonlu sol böbrek mevcuttu (Şekil 1d).
Renalanomali görülmesi sonucu birlikte genital anomali olabileceği düşünülerek yapılan pelvik ultrasonografisinde uterus ve sağ over izlenemedi, sol over 17x6 mm idi. Alt abdomen MR?da uterus band şeklinde ve rudimenter olup sola deviye idi. Sağ overpsoas kası anteriorunda yüksek yerleşimli idi ve 7 mm çapında follikül izlendi. Vajen izlenmedi, fallop tüpleri normaldi. Ayırıcı tanı açısından yapılan kromozom analizi 46, XX olarak sonuçlandı. Hastanın ekokardiyografisinde kardiyak patoloji tespit edilmedi. Hastanın servikotorasik somit displazisi açısından radyolojik görüntülemesi yapıldı, normal olarak raporlandı. Hasta uterus hipoplazisi, ekstremite defekti ve sağ renalagenezi bulguları ile MRKHS tanısı aldı.
Discussion
Bu sendrom ilk olarak Mayer tarafından 1829?da Müllerian agenezi olarak tanımlanmıştır ve daha sonra Rokitansky ve Küster, sendromu; vajina yokluğu, çeşitli uterin anomaliler ile birlikte bilateral normal fonksiyonlu overler, fallop tüpleri ve normal eksternal genitalya olarak tanımlamışlardır 2-5. Hauser renal ve kas-iskelet sistemi anomalilerini de ekleyerek spektrumu genişletmiştir 3. Anomalilerin kapsamı gelişimsel yetmezliğin ne kadar erken ortaya çıktığı ile ilgilidir 4, 6, 7. Strübbe ve arkadaşları, MRKHS?li 91 kadında ürografik, sonografik ve laparoskopik bulguları değerlendirip hastaları Tip A ve Tip B olarak 2 gruba ayırmışlardır 8. İzole uterovajinal aplazi, Rokitansky hastalığı veya Tip A MRKHS olarak tanımlanmıştır. Atipik olarak da adlandırılan Tip B ise daha sık görülmektedir ve ek olarak fallop tüplerinin tek veya çift taraflı displazisi ile renal, kardiyak, ekstremite ve servikotorasik anomaliler eşlik etmektedir. Hastamızda rudimenter uterus, vajen yokluğu tespit edilirken, fallop tüpleri ve overler doğaldı. Literatürde fallop tüplerinin total yokluğu olan vakalar bulunmaktadır 8. Duncan ve arkadaşları, renal ve iskelet sistemi malformasyonları ile ilişkili olduğunda MURCS terimini kullanmışlardır. Müllerian agenezili hastalarda tanımlanan diğer anomaliler, fasial ve işitme defektleri ile çeşitli ekstremite anormallikleridir 9, 10. Prematür adrenarş ile başvuran hastada ekstremite anomalisi olarak sadece sol el birinci karpal kemikte kısalık tespit edildi. Hastaya yapılan işitme testi normaldi.
Müller yapılarında agenezi, gonadal disgeneziden sonra ikinci sıklıkla görülen amenore nedenidir. Rokitansky sendromunda vajinal atrezi, primer amenore, normal vulva, uterus duplikasyon anomalisi, normal dişi genotip ve fenotip, normal overler, renal ve iskelet anomalileri görülebilmektedir. Renal anomaliler olguların %15 ile % 40?ında, en sık skolyoz olmak üzere iskelet anomalileri ise olguların % 30-45?inde görülmektedir 11, 12. Renal ve iskelet anomalileri ile birliktelik her üç sistemin aynı embriyolojik periyotta mezodermal kökenden kaynaklanması sebebiyledir. Hastalar 46 XX karyotipinde ve normal dişi fenotipindedir. Overlerin hormonal fonksiyonu normaldir. Sekonder seks karakterlerinin gelişimi normaldir, fakat menarş gerçekleşmemektedir 8. Normal dişi dış genital yapıya sahip fakat Mülleriyen yapıların (uterus, fallop tüpleri ve üst vajina) bulunmadığı hastalarda bilateral testislerin mevcut olduğu 46, XY cinsiyet gelişim kusuru olasılığı düşünülmelidir. Karyotipi 46, XX saptanan hastada bu olasılık dışlanmıştır.
Genital anormallik renal ve iskelet anomalileri ile birlikte olduğunda MRKHS olarak adlandırılır. Bu anomalilere ek olarak inguinal veya femoral herni, splenozis, TAR trombositopeni, radius yokluğu (TAR) sendromu, anorektal malformasyonlar da literatürde yer almaktadır. Kula ve arkadaşları, MRKHS karakteristikleri arasına konjenital kalp hastalıklarının da eklenmesini önermişlerdir 4.
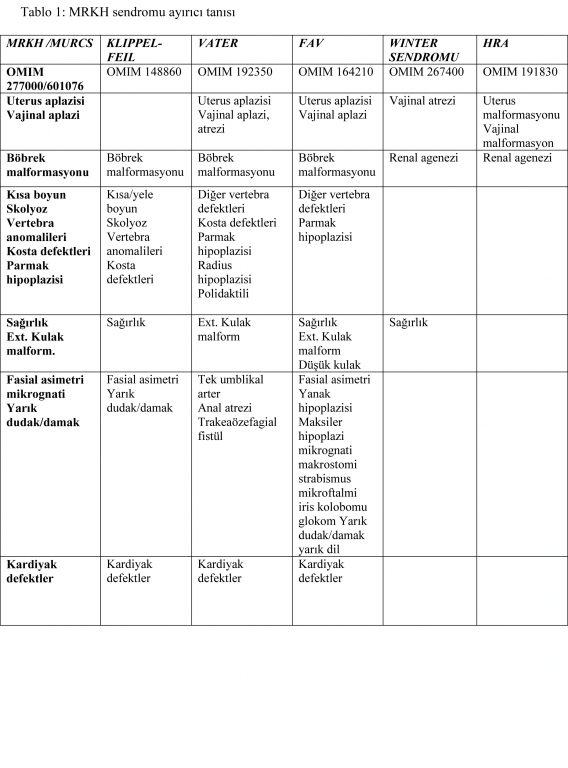
MURCS ayırıcı tanısında vertebral defektler dikkate alındığında Klippel-Feil sendromu ve vertebral defekt, renal agenezi ve uterovajinal displazi açısından ise vertebra anomalileri, anal atrezi fistül, trakeaözafageal fistül, renal anomali (VATER) birlikteliği düşünülmelidir. MURCS?da servikotorasik vertebral defektler beklenirken VATER birlikteliğinde torakalumbar ve sakral vertebralarda defekt gözlenir. MURCS?da gözlediğimiz renal agenezi ve uterovajinal displazi VATER birlikteliğinde nadir gözlenmektedir. VATER birlikteliğinde imperfore anüs, radial displazi gibi üriner ve genital sisteme ait diğer anomaliler ve üst ekstremite anomalileri gözlenir 13. Primer amenorenin önemli bir nedeni olan Turner sendromu da adolesan hastalarda MRKHS?nin ayırıcı tanısında akla getirilmelidir. Hastamızda Turner sendromu dışlanmıştır. Ayırıcı tanı Tablo 1?de özetlenmiştir.
Prematür pubarş ile getirilen, genotip ve fenotip olarak normal kız hastada, parmak anomalisi nedeniyle ileri inceleme yapılarak mülleriyen hipoplazi ve renal agenezi saptanmıştır. MRKHS tanısı alan hasta, erken tanı sayesinde çocuk endokrinoloji ve çocuk nefrolojisi takibine de alınmış, ailesi ileri yaşama yönelik bilgilendirilmiştir. Sonuç olarak, fizik muayenede ekstremite defekti fark edilen kız çocuklarında renal ve genital anomaliler akla getirilmelidir.
References
- Gaughran J. MURCS in a male: a further case. Clin Dysmorphol. 1999; 8:77.
- Russ PD, et al. Mayer-Rokitansky-Kuster-Hauser syndrome diagnosed by magnetic resonance imaging in a 15-year-old girl. J Pediatr Adolesc Gynecol. 1997; 10:89-92.
- Kumar A, Mishra S, Dogra PN. Management of an unusual case of atypical Mayer-Rokitansky-Kuster-Hauser syndrome, with unilateral gonadal agenesis, solitary ectopic pelvic kidney, and pelviureteric junction obstruction. Int Urogynecol J Pelvic Floor Dysfunct. 2007; 18:823-5.
- Wright JE. Failure of mullerian duct development. The Mayer-Rokitansky-Kuster-Hauser syndrome. Aust Paediatr J. 1984; 20:325-7.
- Jurkiewicz B, et al. Rokitansky-Kustner-Hauser syndrome - a case report. Eur J Pediatr Surg. 2006; 16:135-7.
- Kaya H, et al. Mayer-Rokitansky-Küster-Hauser syndrome associated with unilateral gonadal agenesis. A case report. J Reprod Med. 2003; 48:902-4.
- Chapron C, et al. Laparoscopic management of asymmetric Mayer-Rokitansky-Kuster-Hauser syndrome. Hum Reprod. 1995; 10:369-71.
- Strübbe EH, et al. Mayer-Rokitansky-Küster-Hauser syndrome: distinction between two forms based on excretory urographic, sonographic, and laparoscopic findings. AJR Am J Roentgenol. 1993; 160:331-4.
- Ansari MS, Gupta NP, Kriplani A. Ectopic ureter with urinary incontinence. An unusual presentation of Mayer-Rokitansky-Kuster-Hauser syndrome. Int Urogynecol J Pelvic Floor Dysfunct. 2003; 14:64-6.
- Chandiramani M, et al. Mayer - Rokitansky - Kuster - Hauser syndrome. J Obstet Gynaecol. 2006; 26:603-6.
- Strübbe EH, et al. Spinal abnormalities and the atypical form of the Mayer-Rokitansky-Küster-Hauser syndrome. Skeletal Radiol. 1992; 21:459-62.
- Atabek ME, Pirgon O, Sert A. Mayer-Rokitansky-Kuster-Hauser syndrome presenting as premature thelarche in a young child. Pediatr Int. 2007; 49 (4): 533-5.
- Duncan PA, Shapiro LR. MURCS and VATER associations: Vertebral and genitourinary malformations with distinct embryologic pathogenetic mechanisms. Teratology. 1979; 19:24.
|