Abstract
A 23-year-old female patient developed uterine atony after cesarean delivery at term. Thrombocytopenia developed in the postoperative intensive care unit. Peripheral smear revealed 80% neutrophils, 2% monocytes, 18% lymphocytes, erythrocytes polychromasia, diffuse schistosist and 5 normoblast platelets per 100 cells. Diffuse intravascular coagulation, thrombotic thrombocytopenic purpura and hemolytic uremic syndrome were considered. ADAMTS 13 activity level was measured. Her clotting values were normal and she had no clinical signs and symptoms for diffuse intravascular coagulation. When ADAMTS 13 activity level was greater than 10%, the patient was diagnosed as hemolytic uremic syndrome. As soon as the peripheral smear was read, plasmapheresis was started and plasmapheresis was performed 6 times and the patient was discharged with recovery. In this case report, we aimed to present a patient with differential diagnosis of thrombocytopenia caused by pregnancy who developed hemolytic uremic syndrome which was treated effectively.
Keywords :
pregnancy
, thrombocytopenia
, plasmapheresis
Turkish Abstract
Yirmi üç yaşında bayan hastada, miadında sezaryen doğum sonrası, uterin atoni gelişti. Postoperatif yoğun bakım ünitesine alınan hastada trombositopeni gelişti. Periferik yaymada %80 nötrofil, %2 monosit, %18 lenfosit, eritrositlerde polikromazi, yaygın şistozist ve 100 hücreye 5 adet normoblast trombosit görüldü. Hastada, yaygın damar içi pıhtılaşma, trombotik trombositopenik purpura veya hemolitik üremik sendrom gelişmiş olabileceği düşünüldü. Pıhtılaşma değerleri normal olan ve yaygın damar içi pıhtılaşmaya uygun klinik belirtileri olmayan hastanın ADAM-TS 13 aktivite düzeyi incelendi. ADAMTS 13 aktivite düzeyi %10 dan büyük olunca hastaya hemolitik üremik sendrom tanısı konuldu. Periferik yayması okunur okunmaz plazmafereze başlanan hastaya 6 kez plazmaferez yapıldı ve hasta şifa ile taburcu edildi. Biz, bu olgu sunumunda gebeliğin neden olduğu trombositopenilerin ayırıcı tanısını yaptığımız ve hemolitik üremik sendrom tanısı koyup tedavi ettiğimiz bir hastayı sunmayı amaçladık.
Turkish Keywords :
, Gebelik
, trombositopeni
, plazmaferezis
Introduction
Trombositopeni genel olarak trombosit sayısının 150.000 / mm3?in altında olması olarak tanımlanır. 150-100.000 / mm3 arası ılımlı bir düşüşten bahsedilirken 50.000 / mm3?in altı ciddi trombositopeni olarak değerlendirilir. Anemiden sonra en sık görülen ikinci hematolojik bozukluktur 1. Yapılan çalışmalarda gebelikte trombositopeni prevalansının %7-10 arasında olduğunu gösterilmiştir 2-4. Genel olarak, vakaların yaklaşık %75?i gestasyonel trombositopeni (GT), %15-20?si hipertansif bozukluklara sekonder, %3-4?ü immün bozukluklar ve geri kalan %1-2?si nadir diğer durumlar nedeniyle oluşmaktadır 5. Gebeliğe özgü olmayan trombositopeni nedenleri arasında ise, primer immün trombositopeni, viral enfeksiyonlara hepatit B (HBV), sitomegali virus (CMV), Ebstein-Barr virus (EBV)), sistemik lupus eritematozis (SLE) gibi otoimmün hastalıklara ve antifosfolipid antikor sendromuna bağlı gelişen sekonder immün trombositopeni, trombotik mikroanjiyopatiler (trombotik trombositopenik purpura (TTP), hemolitik üremik sendrom (HÜS)), Yaygın damar içi pıhtılaşma (DIC), miyelofibrozis gibi kemik iliği hastalıkları, beslenme ve kullanılan ilaçlar, hipersplenizm ve kalıtsal trombositopeniler sayılabilir 6. Gestasyonel trombositopeni, genellikle son trimesterde ortaya çıkar ve trombosit sayısı tipik olarak 70.000 / mm3 üzerindedir. Doğum sonrası 12 hafta içinde trombosit sayısı normale döner. Fetal ve maternal kanama riski yoktur. Sebebi tam bilinmemekle birlikte, gebelikte oluşan göreceli hemodilüsyon ile beraber plasentada yıkımın artmış olmasıdır 7.
Olgumuzda, miadında gebelik nedeniyle ameliyata alınıp sezaryen sırasında masif kanamayla birlikte uterin atoni gelişen, postoperatif dönemde platelet değerlerinde düşme olması üzerine yoğun bakım ünitesine alınan, gebeliğe bağlı HÜS tanısıyla takip ettiğimiz hastayı sunmayı amaçladık.
Case Report
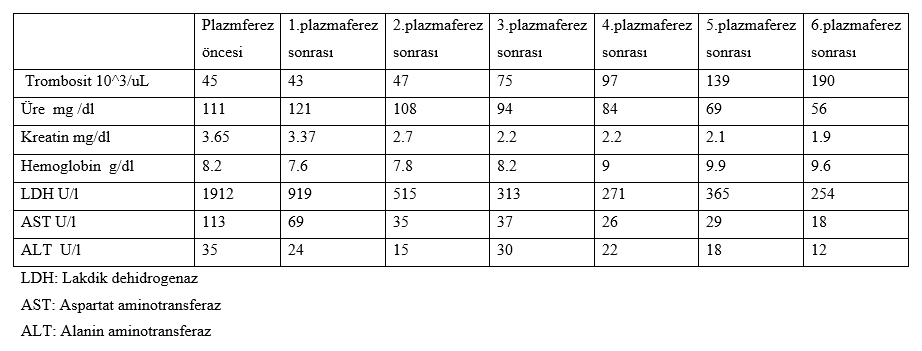
Yirmi üç yaşında bayan hastada miadında sezaryen doğum sonrası uterin atoni gelişti. Uterin atoni nedeniyle hastaya 2 ünite (Ü) eritrosit süspansiyonu (ES), 4 Ü taze donmuş plazma (TDP), 1 Ü trombosit süspansiyon verildi. Hasta postoperatif yoğun bakıma alındı. Tansiyon değerleri ortalama 180/110 mmHg seyreden hastaya perlinganit infüzyonu başlandı. Hastanın trombositleri: 65 000 /mm3, ALT: 37 U/l, AST: 118 U/l, Üre: 91 mg/dl, kreatin: 3.5 mg/dl, Sodyum:133 mmol/L olarak değerlendirildi. İdrar çıkışı mevcut olup saatlik idrar çıkışı takip edildi. Rutin tetkikleri sırasında TSH: 14.54 T4: 0.7 T3: 1.8 olan hastada hipotroidi düşünüldü. Levotiron 1X150 µg başlandı. Yoğun bakımda yatışının 3. gününde hematürisi olan hastanın akciğer grafisinde plevral efüzyon görüldü. Periferik yaymada %80 nötrofil, %2 monosit, %18 lenfosit, eritrositlerde polikromazi, yaygın şistozist ve 100 hücreye 5 adet normoblast trombosit görüldü. Düzeltilmiş retikülosit % 3.2 olarak değerlendirildi. Haptoglubulın 2.8 mg/dl olarak değerlendirildi. DIC?e sekonder trombositopeni, TTP ve HÜS ön tanıları düşünüldü. Hastanın kanama zamanlarının normal olması üzerine DIC tanısı dışlandı. Hematoloji, nefroloji ve yoğun bakım uzmanının ortak görüşü ile tedavi amaçlı plazmaferez yapılmasına karar verildi. Plazmaferez öncesi TTP, HÜS ayrımını yapabilmek için ADAM-TS 13 aktivite düzeyi ölçüldü. Hastanın plazmaferez plazma volumu 3 lt olarak hesaplandı ve 60-70 ml/kg dozunda 6 defa plazmaferez tedavisi uygulandı. Hastaya uygulanan plazmaferz sonrası kan değerlerindeki değişim Tablo 1 de gösterildi.
Plazmaferez tedavisi sonrası hastanın platelet değerlerinde anlamlı artışlar görüldü. 4. plazmaferez tedavisi sonrası hastanın plevral efüzyonunda azalmalar görüldü. Hastanın nörolojık takiplerinde herhangi bir patolojıye rastlanmadı. Plazmaferez öncesi retikülosit sayısı % 3.2 olan hastanın plazmaferez tedavisi sonrası retikülosit sayısı % 9.02 olarak değerlendirildi. Plazmaferez öncesi fibrinojen düzeyi 184 mg/dL olan hastanın plazmaferez sonrası fibrinojen düzeyi:358 mg/dL olarak değerlendirildi. Plazmaferez tedavisine yanıt alınan hastanın ADAMTS 13 düzeyi >10 olunca hastaya HÜS tanısı konuldu. Kan değerleri normalleşen hasta yatışının 15. gününde servise transfer edildi.
Discussion
Gebeliğin neden olduğu trombositopenilerin %2-3?ü immün sistem bozukluklarına sekonder gelişmektedir. Gebeliğin indüklediği trombositopenilerde HÜS-TTP VE DIC ayrımını yapmak bazen zorlaşabilmektedir. Olgumuz 23 yaşında kadın miadında doğum sonrası akut böbrek yetmezliği, hemolitik anemi, trombositopeni, yüksek serum LDH seviyesi ve periferik yaymada parçalanmış eritrositlerin görülmesiyle HÜS-TTP ve DIC ön tanısı almıştır. Hastanın fibrinojen düzeyinin hafif yüksek olması PT, APTT ve INR değerlerinin normal olması nedeni ile DIC tanısı dışlandı 8.
Trombotik trombositopenik purpura ve atipik hemolitik üremik sendrom (aHÜS) aynı organları etkileyebilir ama histopatolojık bulguları farklıdır. Çoğu zaman TTP ve aHÜS?un ayrımı zordur. aHÜS?da daha çok renal bulgular ön planda iken TTP?de santral sinir sistemi bulguları daha belirgindir 9. TTP?nin patogenesinde oluşan abartılı trombosit adezyon ve agregasyonuna ultra large vonWillebrand faktör (ULVWF) denen büyük multimerlerinin neden olduğu gösterilmiştir. Enfeksiyon, gebelik, ilaçlar ve cerrahi ameliyatlar hastalığın ortaya çıkmasına yol açar 10. TTP de oluşan bu abartılı agregasyon ve adezyon beyin damarlarında iskemiye yol açarak nörolojik semptomlara yol açar aynı zamanda renal disfonksiyonlar görülebilir. Hemoliz ve iskemi sonucu serum laktat dehidrogenaz enzim seviyesi artar. Kanama parametreleri genellikle normaldir.
Hemolitik üremik sendrom, mikroanjiyopatik hemolitik anemi, trombositopeni, renal yetmezlik ile karakterize sistemik bir trombotik mikroanjiyopatidir (TMA). Tipik HÜS, Shiga toksin üreten Escherichia coli ya da streptokok enfeksiyonuna bağlı olarak ortaya çıkar. Atipik HÜS aşırı kompleman aktivasyonuna öncülük eden genetik bir mutasyon sonucu oluşan ve yaşamı tehdit eden her yaş grubunda görülebilen kronik bir hastalıktır. AHÜS?ün genetik formunda ADAMTS 13 ü kodlayan genlerde mutasyon mevcuttur 11,12. Atipik HÜS te, düşük trombosit sayısı, hemoliz, beyin, böbrekler, kalp ve diğer organları da etkileyecek şekilde, tüm küçük damarlarda tromboz oluşumu izlenir. Shiga toksin negatiftir. TTP de benzer bulgular görülmekle beraber nörolojik bulgular çoğunlukla izlenir ve atipik HÜS?ün aksine ADAMTS 13 aktivitesi ?5 tir 13. Bu trombosit aktivasyonu ve endotel hasarı, küçük damarlarda çok trombüs oluşumuna, enflamasyon ve oklüzyonla karakterize sistemik TMA?ya neden olur 14,15. Hastaların yarısı erişkin yaş grubundandır. Böbrek hasarı; akut veya kronik renal yetmezlik ya da proteinüri şeklinde kendini gösterebilir. Başlangıç belirtilerinden farklı olarak hastaların çoğunda son dönem böbrek yetmezliği görülebilir.
Atipik HÜS vakaları, serum komleman faktör H, faktör I ve membran faktör proteinindeki MCP gen mutasyonlarıyla birliktedir 16. AHÜS?ün 2010 yılından beri ilk tedavisi plazmaterapidir 17,18,19. Plazmaterapi faktör H, faktör I ve C3 taşır, trombosit hiper agrevasyonunu ve endotel disfonksiyonunu ortadan kaldırır. Plazmaferez renal ve hematolojik fonksiyonlardaki düzelmeyle, volüm yüklenmesi ve kardiyak yetmezliğini de engeller 18. Bizim hastamızda İlk anda kompleman düzeyi ve ADAMTS 13 değeri bilinmediğinden hastadan ADAMATS 13 düzeyi gönderildikten hemen sonra plazmaferez işlemine başlandı. Hastaya 6 gün boyunca günlük 60-70 ml/kg dozundan plazmaferez uygulandı. Trombosit sayısı, LDH ve Hgb düzeyi normale gelene kadar plazmafereze hergün devam edildi. Plazmaferez tedavisi sonrası LDH, trombosit ve üre/cr değerleri normalleşen hastanın ADAMTS 13> 10 olması üzerine hastaya atipik HÜS tanısı kondu.
Sonuç olarak, tedavi edilmediğinde mortalitesi yüksek olan TTP-aHÜS ön tanıları oluşturulduğunda zaman kaybedilmeden hastaya plazmaferez işlemi uygulanmalıdır. Plazmaferez işlemi trombositler ve LDH normalleşene kadar her gün uygulanmalıdır.
References
- Chakraverty R et al. The incidence and cause of coagulopathies in an intensive care population. Br J Haematol. 1996;93(2):460-3.
- Matthews JH et al. Pregnancy-associated thrombocytopenia: definition, incidence and natural history. Acta Haematologica. 1990;84(1):24-9.
- Sainio S et al. Maternal thrombocytopenia at term: a populationbased study. Acta Obstet Gynecol Scand. 2000;79(9):744-9.
- Verdy E et al. Longitudinal analysis of platelet count and volume in normal pregnancy. J Thromb Haemost. 1997;77(4):806-7.
- Burrows RF, Kelton JG. Thrombocytopenia at delivery: a prospective survey of 6715 deliveries. Am Obstet Gynecol. 1990;162(3):731-4.
- McCrae KR. Thrombocytopenia in pregnancy. Hematology Am Soc Hematol Educ Program. 2010;2010(1):397-402.
- Shehata N, Burrows R, Kelton JG. Gestational thrombocytopenia. Clin Obstet Gynecol. 1999;42(2):327-34.
- Carmichae DS, Medley DRK. Heparin in thromboticmicroangiopathy Lancet 1966;1:1421-4.
- Ruggenenti P, Noris M, Remuzzi G. Thrombotic microangiopathy, hemolyticuremic syndrome, and thrombotic thrombocytopenic purpura. Kidney Int. 2001;60(3):831-46.
- Peyvandi F et al. ADAMTS-13 assays in thrombotic thrombocytopenic purpura. J Thromb Haemost. 2010;8(4):631-40.
- Han-MouTsai. Pathophysiology of thromboticthrombocytope-nicpurpura. Int J Hematol. 2010; 91(1):1?19.
- Noris M, Mescia F, Remuzzi G. STEC-HUS, atypi-cal HUS and TTP are all diseases of complement activation. Nat Rev Nephrol . 2012;8 (11):622-33.
- Loirat C, Saland J, Bitzan M. Management of hemolyticuremic syndrome. Presse Med. 2012;41(3 Pt 2): e115-35.
- Giménez Llort A et al. Hemolytic-uraemicsyndrome. A review of 58 cases. An Pediatr (Barc).2008;69(4):297-303.
- Rees L. Atypical HUS: time to take stock of current guidelines and outcome measures? Pediatr Nephrol. 2013;28(5):675-7.
- Ligth C et al. Successful plasma therapy for atypical hemolytic uremic syndrome caused by factor H deficiency owing to a novel mutation in the complement cofactor protein domain 15. Am J Kidney Dis. 2005; 45: 415-21.
- Loirat C, Noris M, Fremeaux-Bacchi V. Complement and the atypical hemolytic uremic syndrome in children. Pediatr Nephrol. 2008;23:1957-72.
- Loirat C et al. Plasmatherapy in atypical hemolytic uremic syndrome. Semin Thromb Hemost. 2010; 36:673-81.
- Noris M et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol. 2010 Oct;5(10):1844-59.
Information Presentation
2019 22.İnternational intencive care symposium da sözlü sunu olarak sunulmuştur
|