Abstract
Rett syndrome is an X-linked dominant neurodevelopmental disorder which is primarily seen in girls.After a normal development for 6-18 months, the disease appears with symptoms such as acquired microcephaly, psychomotor retardation, unpurposeful hand movements. Because of the autistic symptoms commonly seen in the disorder, this syndrome should be considered in the differential diagnosis of many diseases. The definitive diagnosis of Rett syndrome is nowadays with the demonstration of the mutation. However, clinicians need to be familiar with Rett Syndrome diagnostic criteria and the staging of this syndrome in order to request appropriate genetic testing. In this study, we present a 12-year-old female patient with Rett syndrome diagnostic criteria with magnetic resonance imaging findings.
Keywords :
Rett syndrome
, Magnetic resonance imaging
, epileptiform activity
, MECP2 mutation
Turkish Abstract
Rett Sendromu, X?e bağlı dominant kalıtılan, esas olarak kızları etkileyen nörogelişimsel bir hastalıktır. Genellikle doğum ve sonrasındaki 6-18. aylık süreçte normal gelişim gösteren çocuklarda hastalık zamanla gelişen mikrosefali, psikomotor gerilik ve amaçsız el hareketleriyle kendini gösterir. Sendromda otistik bulgulara sıkça rastlanılması bu sendromun birçok hastalığın ayırıcı tanısında akla getirilmesini gerektirmektedir. Rett sendromunun kesin tanısı günümüzde mutasyonun gösterilmesiyle konulmaktadır. Ancak klinisyenlerin genetik incelemeye yönlendirecekleri olguların seçimi için hastalığın tanı kriterlerini ve klinik evrelemesini iyi bilmeleri gerekmektedir. Bu çalışmada Rett sendromu tanı kriterlerinin hepsini içeren 12 yaşında kız hastayı manyetik rezonans görüntüleme bulguları eşliğinde sunuyoruz.
Turkish Keywords :
, Rett sendromu
, Manyetik rezonans görüntüleme
, Epileptiform aktivite
, MECP2 mutasyonu
Introduction
Rett sendromu (RS) normal gelişim basamaklarını takiben erken nörolojik regresyon ile tanınan ve kızlarda görülen nörogelişimsel bir hastalıktır1. Bilişsel, sözel, ince ve kaba motor yetiler ile iletişimin kaybı, otonomik disfonksiyon ve sıklıkla nöbetlerin eşlik ettiği genetik bir hastalıktır. RS kız çocuklarında ağır mental retardasyonun en sık nedenlerinden biridir. RS olanların büyük çoğunluğunda metil-CpG-bağlama proteini-2 (MCEP2) geni mutasyonu mevcuttur. RS?de olgular 6-18 aya kadar normal veya normale yakın gelişim gösterir. Hastaların yaklaşık %1?i ailesel olgulardır ve klinik bulguları olmayan taşıyıcı annelerin çocukları oldukları bildirilmiştir. Biz burada, Rett sendromu tanı kriterlerinin hepsini içeren 12 yaşında kız hastayı manyetik rezonans görüntüleme (MRG) bulguları eşliğinde sunuyoruz.
Case Report
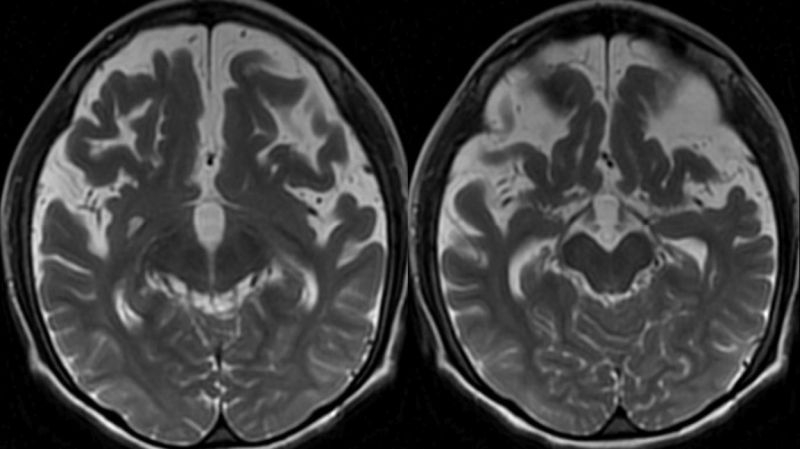
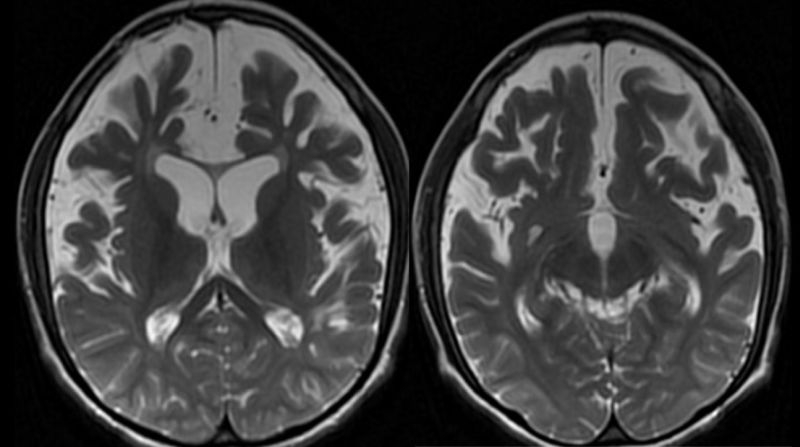
On iki yaşında kız hasta epilepsi, ağır mental-motor retardasyon nedeniyle çocuk nöroloji polikliniğine başvuruyor. Aileden alınan bilgiye göre prenatal ve natal dönemde herhangi bir sorun olmadığı, doğumda baş çevresinin normal olduğu, erken motor ve mental gelişim basamaklarının normal olduğu ve anne ve baba arasında akrabalık olmadığı öğrenildi. Hasta 12 yaşında ve 20 kg ağırlığındaydı. Hastanın fizik muayenesinde baş çevresinin 49,5 cm (2 SD altında) olarak ölçüldü. Nörolojik muayenesinde mikrosefali dışında steriotipik el hareketleri, otistik bulguları ve mental-motor gelişme geriliği olan hastanın ataksik geniş kaideli yürüyüşü vardı. Hastanın, rutin hematoloji, biyokimya ve idrar testleri ile doğumsal metabolik hastalık tarama testleri normal sınırlarda idi. Yapılan elektroensefalografi (EEG)?de jeneralize tipte epileptiform bulgular saptandı. Kraniyal magnetik rezonans görüntüleme (MRG)?de; korpus kallozumda incelme ve prepontin sisternde genişleme (Şekil 1), frontal ve temporal loblarda belirgin atrofi ve bu düzeyde subkortikal-periventriküler beyaz cevher hacminde azalma izlendi (Şekil 2).
Lateral ve üçüncü ventrikülde ve Silvian fissurde dilatasyon vardı (Şekil 3).
Bilateral periventriküler ve subkortikal beyaz cevherde, sentrum semiovalede, eksternal kapsül ve insular lobda T2A ve FLAIR sekanslarda hiperintens sinyal değişiklikleri dikkati çekti. Olgunun moleküler incelemesinde MCEP2 geninin 3. Ekzonunda de novo mutasyon saptandı. RETT sendromu tanısı konan olguya antiepileptik başlandı ve takibe alındı.
Discussion
Rett sendromu ilk defa 1966 yılında Andreas Rett tarafından tanımlanan, sıklıkla kız çocuklarında görülen nörolojik bir hastalıktır 1. Klinik özellikleri arasında; otistik davranış, mental retardasyon, solunumsal bozukluklar, konuşma yeteneğinin ve el becerilerinin kaybı, ellerini amaçlı kullanamama, nefes tutma, oral-motor disfonksiyonlar, gastrointestinal motilite bozuklukları, skolyoz, otonomik disfonksiyon, ve somatik gelişme bozuklukları mevcuttur 2. Tanı X kromozomu üzerinde bulunan MECP2 geninin kusurlu olduğunun gösterilmesiyle konur 2.
Dört klinik evre süreci vardır. Birinci evre; 6-18 aylar arasında gelişme geriliğinin başlama evresidir. İkinci evre; 1-4 yaş arası edinilmiş beceriler ve mental kayıp evresidir. Üçüncü evre; 4-10 yaş arası kabul edilebilir hareketlerle birlikte nöromotor gerilik bulunur. Dördüncü evrede ise; ciddi bir yetersizlik olup, tekerlekli sandalyeye bağımlılık gelişir. Bütün semptomlar tedaviye dirençsiz bir hal alır 2-4. Hastamız dördüncü evrededir. Tanı için destekleyici kriterler arasında belirtilen epilepsi nöbetlerinin RS?li olgularda %50?90 oranında görüldüğü bilinmektedir 5.
RS li hastalarda yaşla birlikte artış gösteren beyinde global hipoplazi ve progressif serebellar atrofi izlenmiştir 6. Bununla birlikte kortikal atrofinin derecesi yaştan ziyade sendromun ciddiyeti ile ilişkilidir. Beyin hacmindeki azalmanın progressif olmadığı gösterilmiştir 7. RS?li olgularda beyin sapının daraldığı bildirilmiştir 8. Ayrıca otistik olgularda beyin sapı, serebeller vermis ve korpus kallozum normalden küçüktür 9,10.
Sonuç
Rett sendromunun kesin tanısı günümüzde mutasyonun gösterilmesiyle konulmaktadır. Kızlarda görülen, ilerleyici nörogelişimsel bir bozukluktur .Genetik boyutun yanında eşlik eden MRG görüntüleme bulgularını bilmek hastalığı tanımada ve erken tedavi yolunu belirlemede kolaylık sağlayabilir.
References
- Armstrog DD. Review of Rett Seyndrome. J Neuropathol Exp Neurol. 1997; 56: 843-9.
- Huppke P, et al. Rett Syndrome: analysis of MECPP2 and clinical characterization of 31 patients. Hum Mol Genet. 2000; 9:1369-75.
- Haliloğlu G, Topçu M Ağaoğlu B. Rett Sendromu. Çetin FÇ, Coşkun A, İşeri E, Miral S, Motavallı N, Pehlivantürk B, Türkbay T, Uslu R, Ünal F, editörler. Çocuk ve Ergen Psikiyatrisi Temel Kitabı. Birinci Baskı. Ankara: Hekimler Yayın Birliği; 2008: 264-274.
- Aras Ş. Rett sendromu: klinik özellikler ve genetik etiyoloji. Demans Dergisi. 2003; 3:93-9.
- Nissenkorn A, et al. Epilepsy in Rett syndrome-the experience of a National Rett Center. Epilepsia. 2010; 51:1252-8.
- Murakami JW, et al. Cerebellar and cerebral abnormalities in Rett syndrome: a quantitative MR analysis. Am J Roentgenol. 1992;159:177-83.
- Vanhala R, Gaily E, Paetau A, Riikonen R. Pons tumour behind a phenotypic Rett syndrome presentation. Dev Med Child Neurol. 1998; 40:836-39.
- Nihei K, Naitoh H. Cranial computed tomographic and magnetic resonance imaging studies on the Rett syndrome. Brain Dev. 1990;12:101-5.
- Hashimoto T, Tayama M, Miyazaki M, Murakawa K, Kuroda Y. Brainstem and cerebellar vermis involvement in autistic children. J Child Neurol 1993; 8:149-153.
- Manes F, et al. . An MRI study of the corpus callosum and cerebellum in mentally retarded autistic individuals. J Neuropsychiatry Clin Neurosci. 1999; 11:470-4.
Information Presentation
Türk Manyetik Rezonans Derneği 24. Yıllık Uluslararası Katılımlı Bilimsel Toplantısında poster olarak sunulmuştur.
|