Abstract
Crouzon syndrome, also known as craniofacial dysostosis, is an autosomal dominant congenital syndrome that affects the skull and facial bones. Genetic defect resulting from FGFR-2(fibroblast growth factor receptor 2) gene mutation is the clinical result. Craniosynostosis, maxillar hypoplasia, shallow orbit, ocular proptosis and hypertelorism are characteristic features of Crouzon syndrome. In this study, a case with Crouzon syndrome and multiple anomalies was presented because of its rarity.
Keywords :
Anomaly
, Crouzon syndrome
, Gene mutation
, Craniosinocytosis
Turkish Abstract
Kraniofasial disostosis olarak da bilinen Crouzon sendromu, kafatası ve yüz kemiklerinin etkilendiği otozomal dominant geçişli konjenital bir sendromdur. Fibroblast büyüme faktörü reseptör -2 (FGFR-2 ) gen mutasyonuna bağlı genetik defekt sonucu klinik oluşmaktadır. Kraniosinostoz, maksillar hipoplazi, sığ orbita, oküler proptozis ve hipertelorizm, Crouzon sendromunun karakteristik özellikleridir. Bu çalışmada, Crouzon sendromu tanısı alan ve multiple anomalileri olan bir olgu, nadir görülmesi nedeniyle sunuldu.
Turkish Keywords :
, Anomali
, Crouzon sendromu
, Gen mutasyonu
, Kraniosinositoz
Introduction
Crouzon sendromu otozomal dominant geçişli, kraniyosinostoz ve dismorfik yüz görünümü ile karakterize olan bir sendromdur. Dismorfik özelliklerin derecesine göre yenidoğan veya infant döneminde tanı konur. Otozomal dominant geçiş gösterip, görülme sıklığı ırk, bölge ve etnik kökene göre değişiklik göstermekte olup ortalama 16/1.000.000 olan genetik bir hastalıktır 1. Klinik fibroblast büyüme faktörü reseptör-2 (FGFR-2)?deki mutasyonlar sonucu ortaya çıkmaktadır 2. Crouzon sendromunda özellikle koranal sütürlerin ve sagittal sütürün erken kapanması sonucu akrosefali veya brakisefali görülebilir. Ayrıca, hastalarda belirgin ekzoftalmus, pitozis, hipertelorizm, gaga şeklinde burun, kulak ve damak anomalileri mevcut olabilir 3.
Crouzon sendromlu hastalarda çok sayıda anomali birlikteliği olabileceğinden bu hastalar dikkatli ve detaylı muayene edilmelidirler. Bu çalışmada, Crouzon sendromu tanısı alan ve farklı anomalilere sahip bir olgu sunuldu.
Case Report
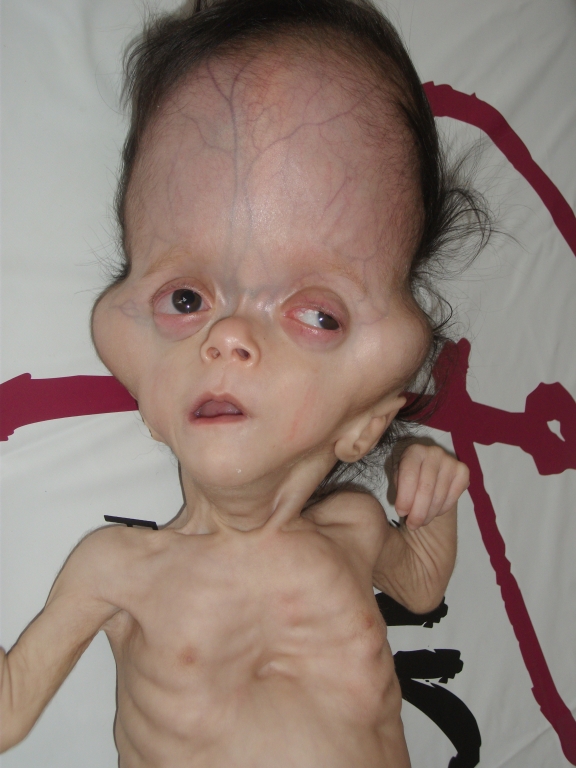
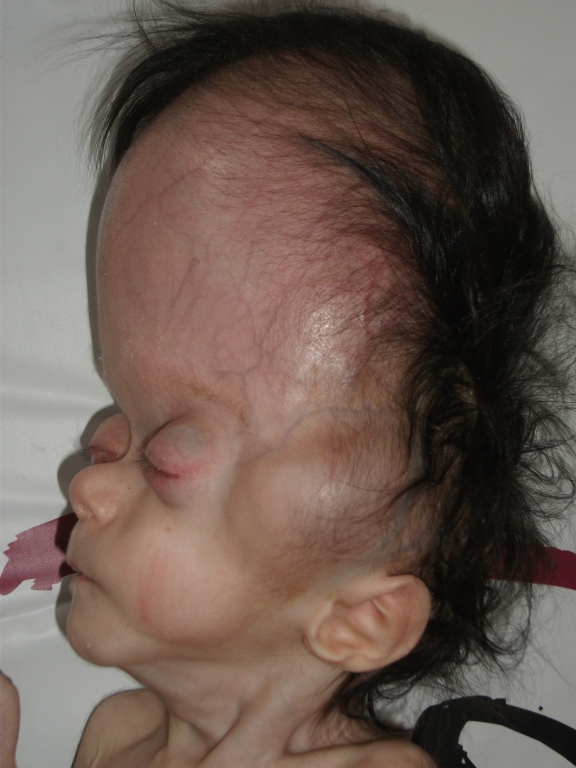
Beş aylık kız hasta baş, yüz ve gözlerde mevcut olan şekil bozukluğu nedeniyle kliniğimize getirildi. Hastanın öz ve soy geçmişinde özellik saptanmadı. Yapılan fizik muayenesinde vücut ağırlığı 3500 gram (<3p), boy 62 cm (3-10p), baş çevresi 36 cm (<3p) olarak bulundu. Baş mikrosefalik görünümde, sagittal sütür separe, ön fontanel 4x4 cm ve arka fontanel 3x3 cm açıklıkta idi. Ayrıca, hastada brakisefali, düşük kulak, düz alın, gaga şeklinde burun ve burun kökü basıklığı, hipoplazik maksilla ve yüksek damak anomalisi varlığı saptandı (Şekil 1A, Şekil 1B). Belirgin ekzoftalmus ve sol gözde ekzotropiası vardı. Ekstremite muayenesinde yaygın hipotoni ve ellerde kortikal fisting bulgusu mevcuttu.
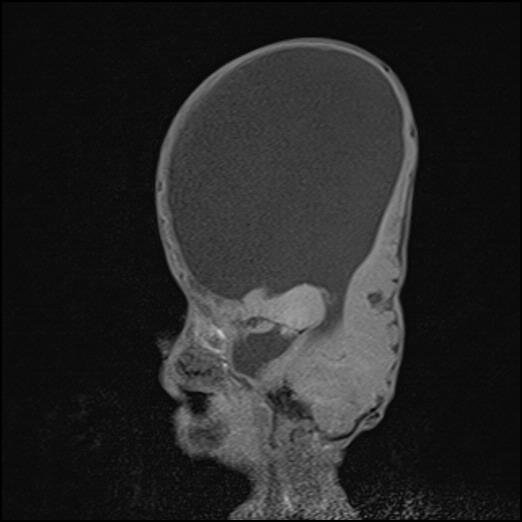
Pektus ekskavatusunun olduğu gözlenen olguda solunum ve kalp sesleri normal olup ek ses veya üfürüm duyulmadı. Batın normal bombelikte idi, organomegali saptanmadı. Nörolojik muayenesinde göz kontağı kurulamayan hastanın baş kontrolü yoktu. Belirgin turunkal hipotonisi mevcuttu. Laboratuvar bulgularından hemogram, tam idrar tahlili, kan şekeri, serum elektrolitleri, karaciğer ve böbrek fonksiyon testleri normal bulundu. İki yönlü kraniografide koronal sütürlerde sinostoz saptandı. PA akciğer grafisi, uzun kemik grafileri, vertebra grafisi ve batın ultrasonografisi normal olarak değerlendirildi. Kranial tomografide kranium kemikleri asimetrik olup, düzensiz kontur ve kalınlık göstermekteydi, üçüncü ventrikül ve lateral ventriküller dilate, posterior fossa ve beyin parenkimi basılı görünümde idi. Beyin manyetik rezonans görüntülemede korpus kallosum agenezisi mevcuttu. Posterior fossa normalden küçük idi, üçüncü ventrikül ve lateral ventriküller de ileri derecede dilatasyon izlendi (Şekil 2).
Discussion
Crouzon sendromu, ilk kez 1912 yılında Crouzon tarafından kraniosinostoz, sindaktili, derin olmayan sığ orbita ve maksillar hipoplazi ile karakterize bir hastalık olarak tanımlanmıştır. Reardan ve arkadaşları FGFR-2 genindeki mutasyonların bu sendroma neden olduğunu bildirmiştir. Bununla birlikte, yeni mutasyonların ilerlemiş anne yaşı ile ilgili olduğu rapor edilmiştir 3.
Bu sendromda temel olarak, koronal sütürlerin ve sagittal sütürlerin prematür füzyonu sonucu akrosefali, brakisefali, çıkık alın ve düzleşmiş oksiput görülür. Belirgin ekzoftalmus, pitozis, hipertelorizm, gaga şeklinde burun, şaşılık, kısa üst dudak, göreceli prognatizm, kulak ve damak deformiteleri meydana gelir 4. Olguların %80?inde optik atrofi veya papil ödem mevcuttur ve muhtemelen artmış kafa içi basıncına bağlıdır. Hastalarda % 92 oranında şaşılık görülür ve en sık görülen patern ekzotropya şeklindedir 5.
Bizim hastamızın anne yaşı 38 idi ve kromozom incelemesi normaldi. Hastamızın muayenesinde akrosefali, yüksek alın, düzleşmiş oksiput, küçük ağız, maksiller hipoplazi, hipertelorizm, relatif mandibular prognatizm ve belirgin eksoftalmus vardı. Ayrıca, muayenede sol gözde ekzotropiasının olduğu tespit edildi (Şekil 1A).
Crouzon sendromunda iki veya daha fazla sütürün tutulması intrakraniyal basınç artımına neden olmaktadır. Bu olgularda, beyinin gelişmesi ile kafatası kemik yapıları arasındaki uyumsuzluk sonucu hidrosefali gelişme riski daha yüksektir. Motor ve mental retardasyon derecesi hastalara göre değişiklik gösterir. Olgularda mental retardasyon gelişme sıklığı %1 ile 20 arasında bildirilmiştir. Kardiyovasküler malformasyon görülme sıklığı ise oldukça yüksek bulunmuştur 6.
Baş kontrolü olmayan ve göz kontağı kurulamayan hastamızda belirgin derecede nöromotor gelişim geriliği mevcuttu. Beyin manyetik rezonans görüntülemede, posterior fossa normalden küçük, üçüncü ventrikül ve lateral ventriküller de ileri derecede dilatasyon vardı. Ayrıca, korpus kallosum agenezisi mevcuttu. Ekografisinde patent duktus arteriosus tespit edildi.
Tanı genellikle mevcut fenotipik özellikleri olan hastalarda çekilen kraniografide sagittal ve koronal sütürlerde sinostosiz görülmesi ile konur. Hastalığın kesin tedavisi yoktur. Kraniosyonostoz için kraniotomi ve fasial deformiteler için kozmetik düzeltme ameliyatları yapılabilir 7,8.
Sonuç olarak, Crouzon sendromu tanısı alan hastalar dikkatli ve detaylı muayene edilmelidirler. Bu olgularda farklı anomalilerin olabileceği düşünülmelidir. Ayrıca, hastaların multidisipliner takip ve tedavilerinin gerekliliği unutulmamalıdır.
Acknowledgement
Tüm yazarlar, veri analizine katkıda bulunmuş, kağıda ilişkin taslak hazırlama ve gözden geçirme çalışmalarına katkıda bulunmuş ve çalışmaların her alanında hesap verebilir olmayı kabul etmişlerdir. Bu olgu sunumunda herhangi bir çıkar çatışması yoktur.
References
- Lin Y, et al. FGFR2 mutations and associated clinical observations in two Chinese patients with Crouzon syndrome. Mol Med Rep. 2017 ; 16: 5841-46.
- Ahmed I, Afzal A. Diagnosis and evaluation of Crouzon syndrome. J Coll Physicians Surg Pak. 2009; 19: 318-20.
- T. Maeda, et al. Clinically mild, atypical, and aged craniofacial syndrome is diagnosed as Crouzon syndrome by identification of a point mutation in the fibroblast growth factor receptor 2 gene (FGFR2). Internal Medicine. 2004;43 (5): 432-5.
- Kaushik A, et al. Crouzon's Syndrome: A Rare Genetic Disorder. Int J Clin Pediatr Dent. 2016;9(4):384-7.
- Qiao T, et al. Surgical Treatment of V-pattern Exotropia in Crouzon Syndrome. J Pediatr Ophthalmol Strabismus. 2015;52(5):299?304.
- Priyadharshini R, et al. A cross-sectional study of cardiac anomalies among children with orofacial cleft-role of echocardiography. Int J Contemp Pediatr. 2017;4(4):1274-77
- Renier D, et al. Management of craniosynostoses. Childs Nerv Syst. 2000;16: 645-58.
- Driessen C, et al. he effect of early fusion of the spheno-occipital synchondrosis on midface hypoplasia and obstructive sleep apnea in patients with Crouzon syndrome. J Craniomaxillofac Surg. 2017;45(7):1069-73.
|